Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine Learning Harnesses Molecular Dynamics to Discover New $μ$ Opioid Chemotypes
Paper and Code
Mar 12, 2018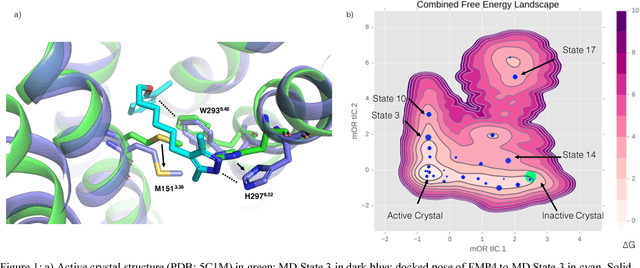
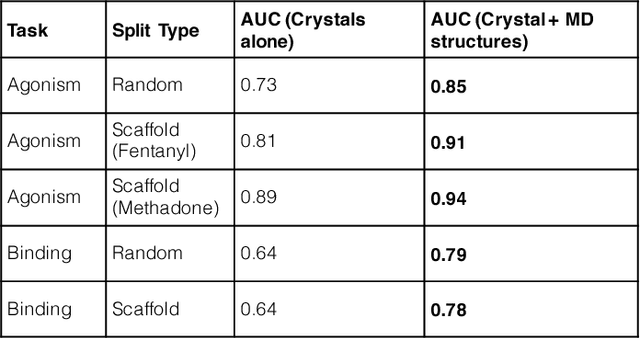
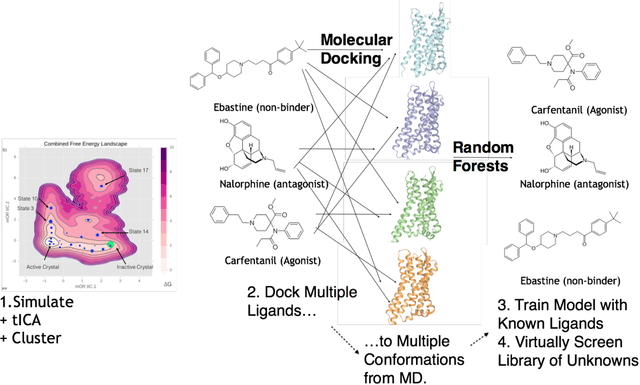
Computational chemists typically assay drug candidates by virtually screening compounds against crystal structures of a protein despite the fact that some targets, like the $\mu$ Opioid Receptor and other members of the GPCR family, traverse many non-crystallographic states. We discover new conformational states of $\mu OR$ with molecular dynamics simulation and then machine learn ligand-structure relationships to predict opioid ligand function. These artificial intelligence models identified a novel $\mu$ opioid chemotype.
* 28 pages, machine learning, computational biology, GPCRs, molecular
dynamics, molecular docking, molecular simulation
View paper on