 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials
Oct 04, 2023



Machine learning plays an important and growing role in molecular simulation. The newest version of the OpenMM molecular dynamics toolkit introduces new features to support the use of machine learning potentials. Arbitrary PyTorch models can be added to a simulation and used to compute forces and energy. A higher-level interface allows users to easily model their molecules of interest with general purpose, pretrained potential functions. A collection of optimized CUDA kernels and custom PyTorch operations greatly improves the speed of simulations. We demonstrate these features on simulations of cyclin-dependent kinase 8 (CDK8) and the green fluorescent protein (GFP) chromophore in water. Taken together, these features make it practical to use machine learning to improve the accuracy of simulations at only a modest increase in cost.
Step Change Improvement in ADMET Prediction with PotentialNet Deep Featurization
Mar 28, 2019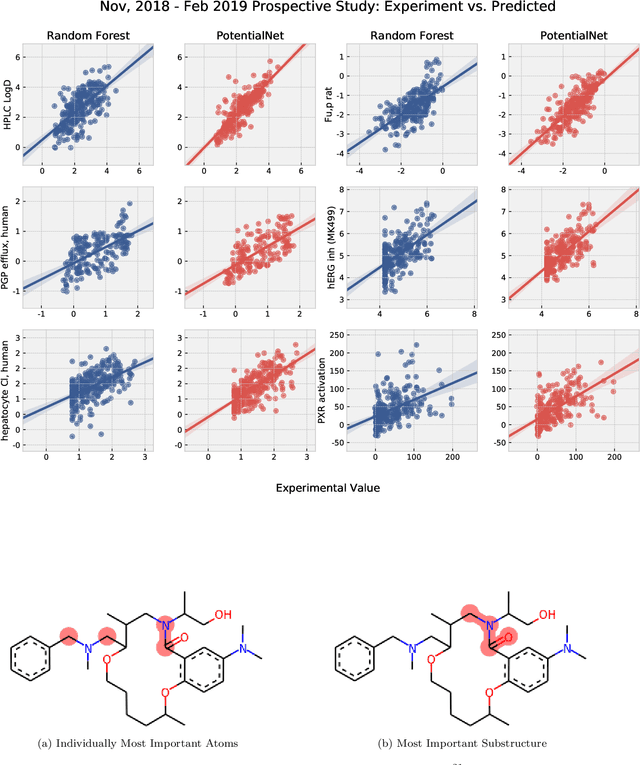
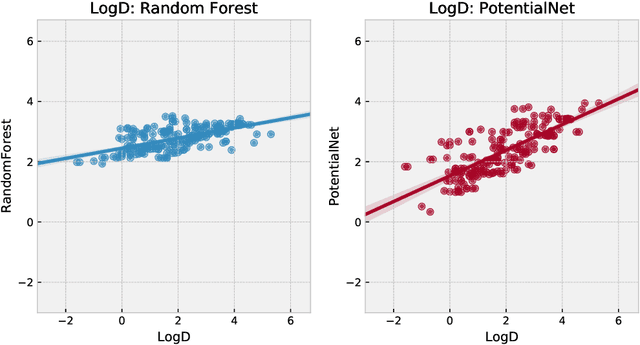
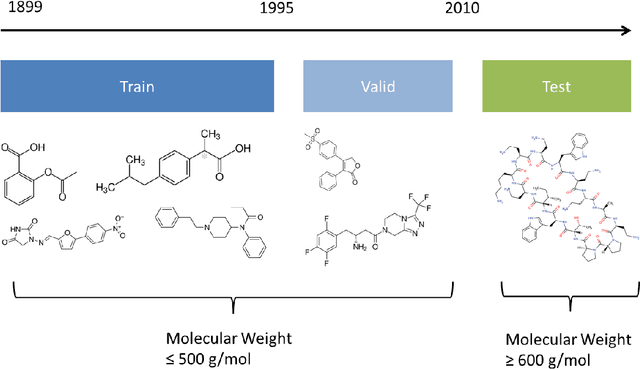
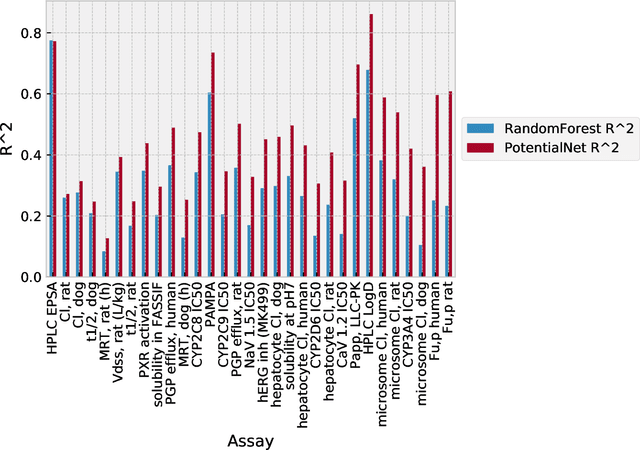
The Absorption, Distribution, Metabolism, Elimination, and Toxicity (ADMET) properties of drug candidates are estimated to account for up to 50% of all clinical trial failures. Predicting ADMET properties has therefore been of great interest to the cheminformatics and medicinal chemistry communities in recent decades. Traditional cheminformatics approaches, whether the learner is a random forest or a deep neural network, leverage fixed fingerprint feature representations of molecules. In contrast, in this paper, we learn the features most relevant to each chemical task at hand by representing each molecule explicitly as a graph, where each node is an atom and each edge is a bond. By applying graph convolutions to this explicit molecular representation, we achieve, to our knowledge, unprecedented accuracy in prediction of ADMET properties. By challenging our methodology with rigorous cross-validation procedures and prospective analyses, we show that deep featurization better enables molecular predictors to not only interpolate but also extrapolate to new regions of chemical space.
PotentialNet for Molecular Property Prediction
Oct 22, 2018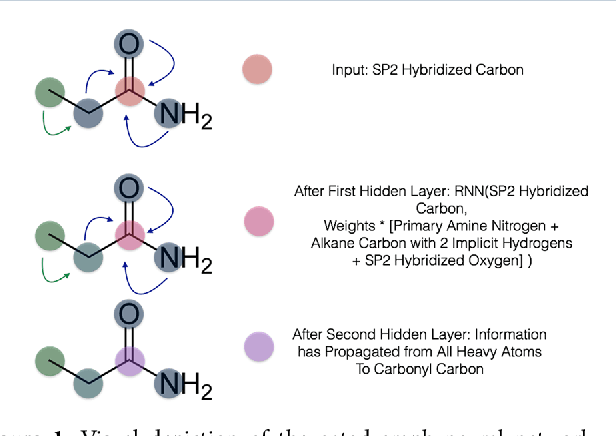
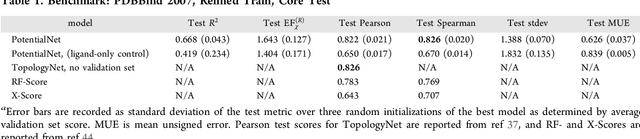
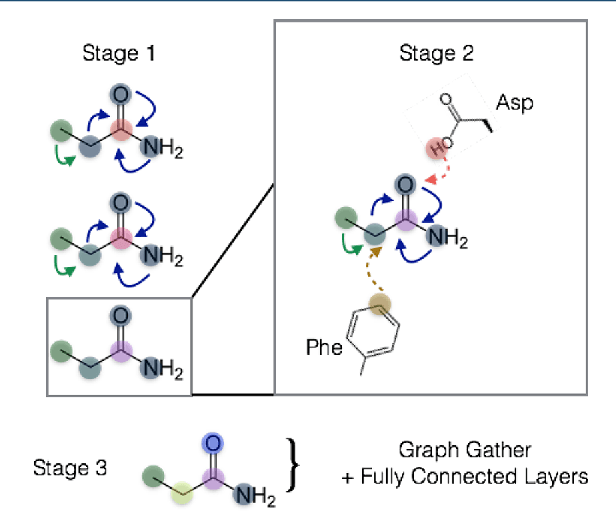
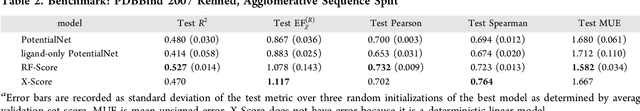
The arc of drug discovery entails a multiparameter optimization problem spanning vast length scales. They key parameters range from solubility (angstroms) to protein-ligand binding (nanometers) to in vivo toxicity (meters). Through feature learning---instead of feature engineering---deep neural networks promise to outperform both traditional physics-based and knowledge-based machine learning models for predicting molecular properties pertinent to drug discovery. To this end, we present the PotentialNet family of graph convolutions. These models are specifically designed for and achieve state-of-the-art performance for protein-ligand binding affinity. We further validate these deep neural networks by setting new standards of performance in several ligand-based tasks. In parallel, we introduce a new metric, the Regression Enrichment Factor $EF_\chi^{(R)}$, to measure the early enrichment of computational models for chemical data. Finally, we introduce a cross-validation strategy based on structural homology clustering that can more accurately measure model generalizability, which crucially distinguishes the aims of machine learning for drug discovery from standard machine learning tasks.
Automated design of collective variables using supervised machine learning
May 13, 2018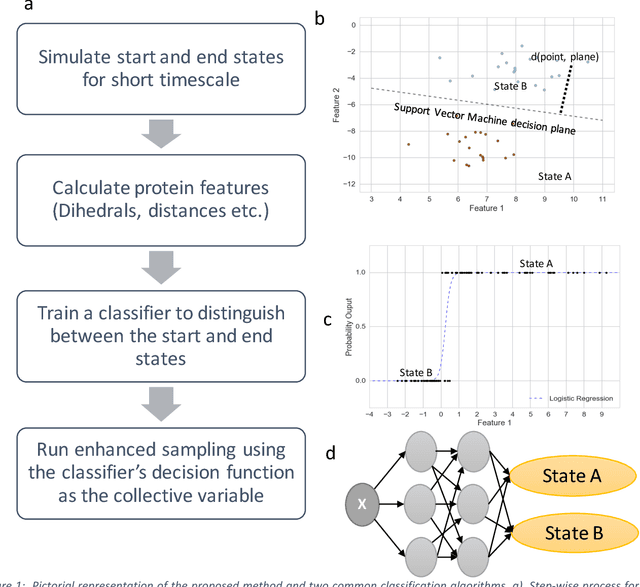
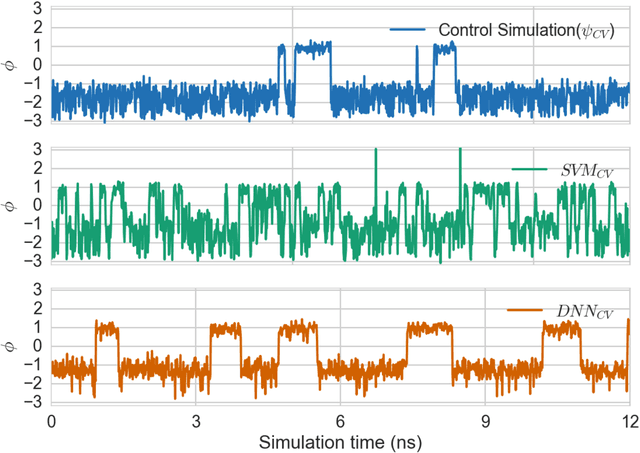
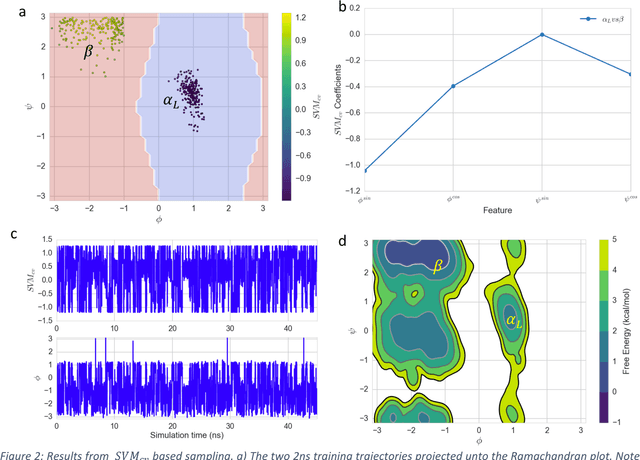

Selection of appropriate collective variables for enhancing sampling of molecular simulations remains an unsolved problem in computational biophysics. In particular, picking initial collective variables (CVs) is particularly challenging in higher dimensions. Which atomic coordinates or transforms there of from a list of thousands should one pick for enhanced sampling runs? How does a modeler even begin to pick starting coordinates for investigation? This remains true even in the case of simple two state systems and only increases in difficulty for multi-state systems. In this work, we solve the initial CV problem using a data-driven approach inspired by the filed of supervised machine learning. In particular, we show how the decision functions in supervised machine learning (SML) algorithms can be used as initial CVs (SML_cv) for accelerated sampling. Using solvated alanine dipeptide and Chignolin mini-protein as our test cases, we illustrate how the distance to the Support Vector Machines' decision hyperplane, the output probability estimates from Logistic Regression, the outputs from deep neural network classifiers, and other classifiers may be used to reversibly sample slow structural transitions. We discuss the utility of other SML algorithms that might be useful for identifying CVs for accelerating molecular simulations.
Deep Learning Phase Segregation
Mar 23, 2018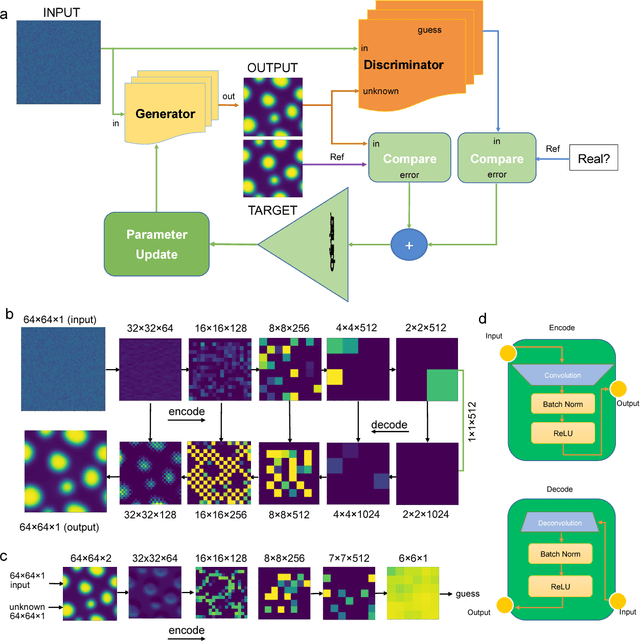
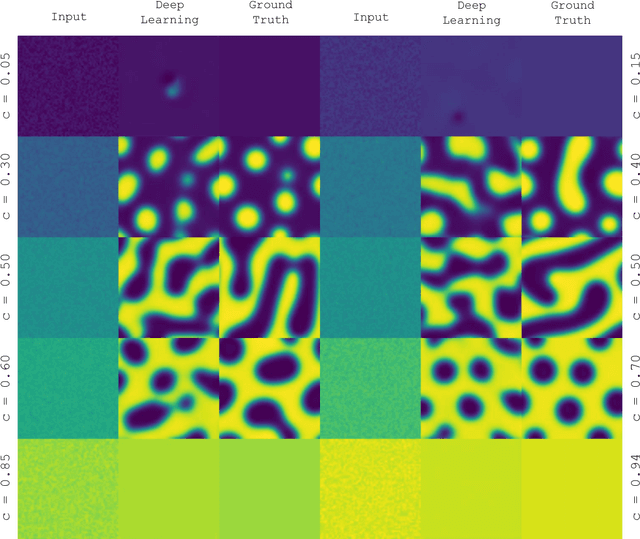


Phase segregation, the process by which the components of a binary mixture spontaneously separate, is a key process in the evolution and design of many chemical, mechanical, and biological systems. In this work, we present a data-driven approach for the learning, modeling, and prediction of phase segregation. A direct mapping between an initially dispersed, immiscible binary fluid and the equilibrium concentration field is learned by conditional generative convolutional neural networks. Concentration field predictions by the deep learning model conserve phase fraction, correctly predict phase transition, and reproduce area, perimeter, and total free energy distributions up to 98% accuracy.
Note: Variational Encoding of Protein Dynamics Benefits from Maximizing Latent Autocorrelation
Mar 17, 2018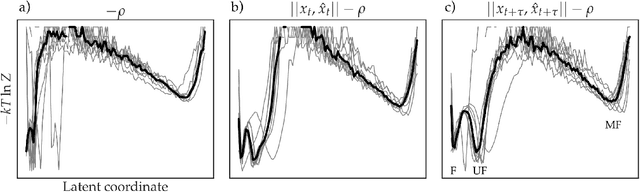
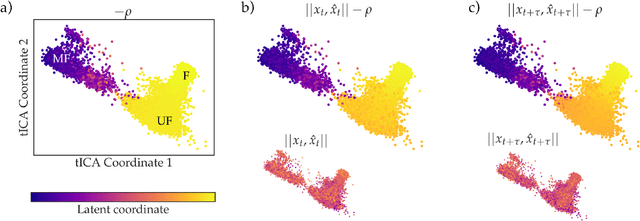
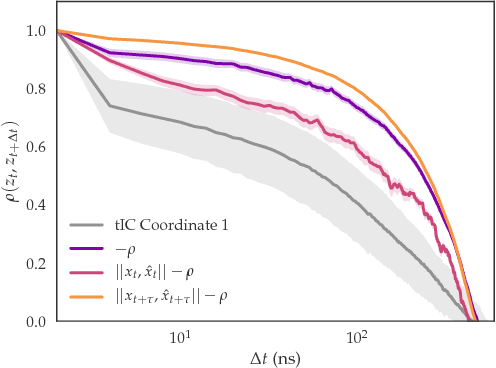
As deep Variational Auto-Encoder (VAE) frameworks become more widely used for modeling biomolecular simulation data, we emphasize the capability of the VAE architecture to concurrently maximize the timescale of the latent space while inferring a reduced coordinate, which assists in finding slow processes as according to the variational approach to conformational dynamics. We additionally provide evidence that the VDE framework (Hern\'andez et al., 2017), which uses this autocorrelation loss along with a time-lagged reconstruction loss, obtains a variationally optimized latent coordinate in comparison with related loss functions. We thus recommend leveraging the autocorrelation of the latent space while training neural network models of biomolecular simulation data to better represent slow processes.
Machine Learning Harnesses Molecular Dynamics to Discover New $μ$ Opioid Chemotypes
Mar 12, 2018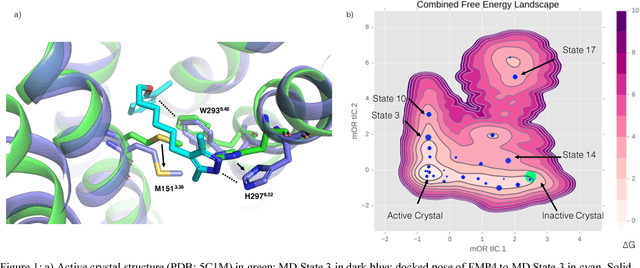
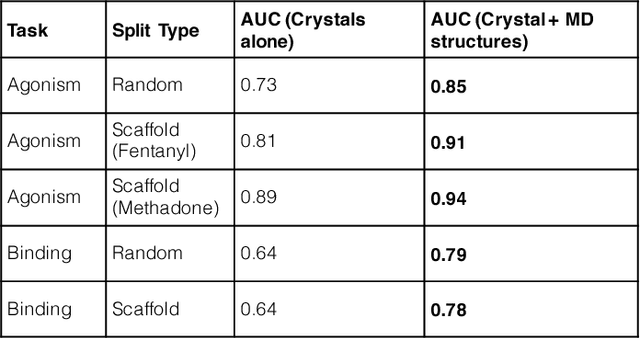
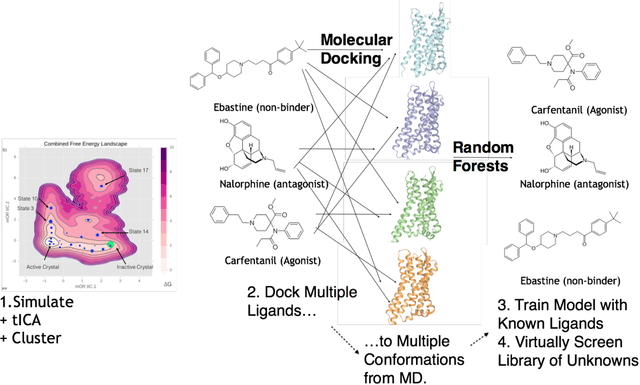
Computational chemists typically assay drug candidates by virtually screening compounds against crystal structures of a protein despite the fact that some targets, like the $\mu$ Opioid Receptor and other members of the GPCR family, traverse many non-crystallographic states. We discover new conformational states of $\mu OR$ with molecular dynamics simulation and then machine learn ligand-structure relationships to predict opioid ligand function. These artificial intelligence models identified a novel $\mu$ opioid chemotype.
SentRNA: Improving computational RNA design by incorporating a prior of human design strategies
Mar 08, 2018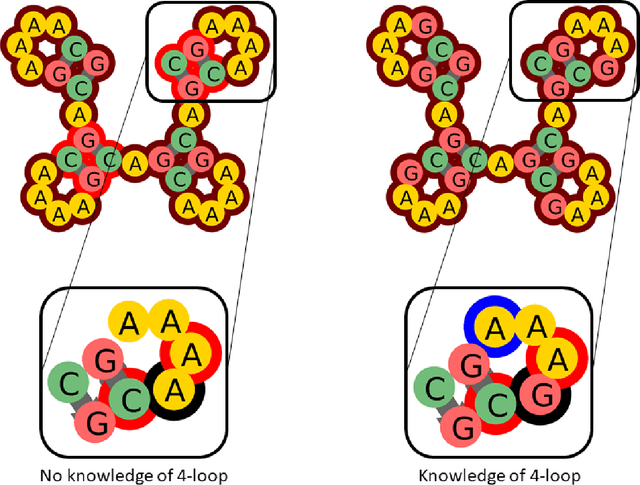
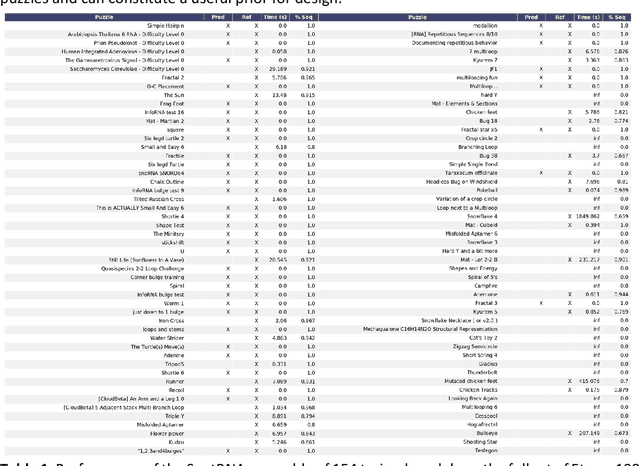
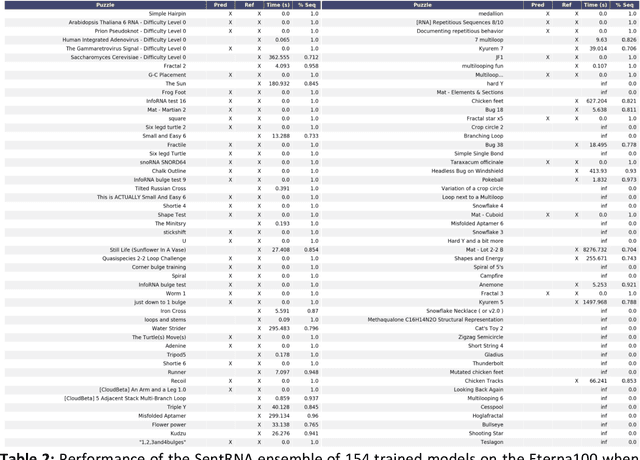
Designing RNA sequences that fold into specific structures and perform desired biological functions is an emerging field in bioengineering with broad applications from intracellular chemical catalysis to cancer therapy via selective gene silencing. Effective RNA design requires first solving the inverse folding problem: given a target structure, propose a sequence that folds into that structure. Although significant progress has been made in developing computational algorithms for this purpose, current approaches are ineffective at designing sequences for complex targets, limiting their utility in real-world applications. However, an alternative that has shown significantly higher performance are human players of the online RNA design game EteRNA. Through many rounds of gameplay, these players have developed a collective library of "human" rules and strategies for RNA design that have proven to be more effective than current computational approaches, especially for complex targets. Here, we present an RNA design agent, SentRNA, which consists of a fully-connected neural network trained using the $eternasolves$ dataset, a set of $1.8 x 10^4$ player-submitted sequences across 724 unique targets. The agent first predicts an initial sequence for a target using the trained network, and then refines that solution if necessary using a short adaptive walk utilizing a canon of standard design moves. Through this approach, we observe SentRNA can learn and apply human-like design strategies to solve several complex targets previously unsolvable by any computational approach. We thus demonstrate that incorporating a prior of human design strategies into a computational agent can significantly boost its performance, and suggests a new paradigm for machine-based RNA design.
Using Deep Learning for Segmentation and Counting within Microscopy Data
Feb 28, 2018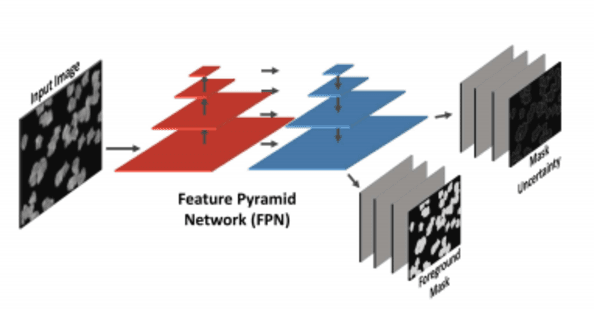
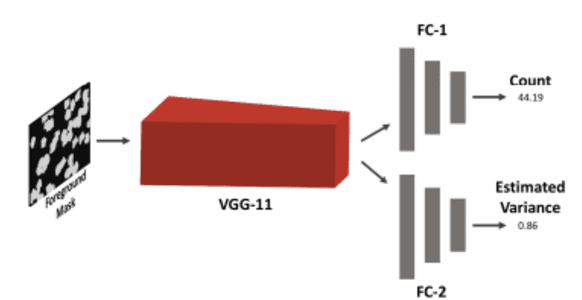
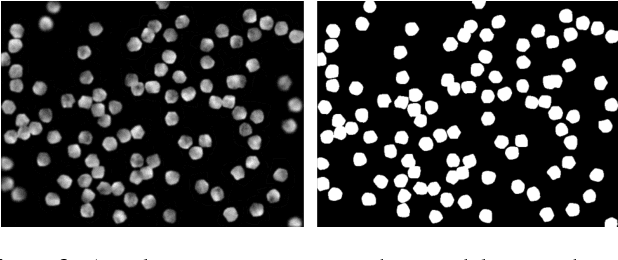

Cell counting is a ubiquitous, yet tedious task that would greatly benefit from automation. From basic biological questions to clinical trials, cell counts provide key quantitative feedback that drive research. Unfortunately, cell counting is most commonly a manual task and can be time-intensive. The task is made even more difficult due to overlapping cells, existence of multiple focal planes, and poor imaging quality, among other factors. Here, we describe a convolutional neural network approach, using a recently described feature pyramid network combined with a VGG-style neural network, for segmenting and subsequent counting of cells in a given microscopy image.
Transferable neural networks for enhanced sampling of protein dynamics
Jan 02, 2018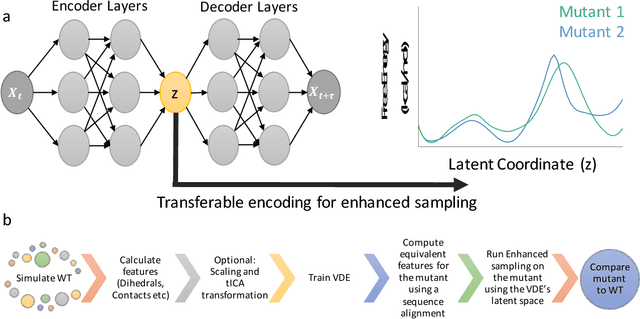
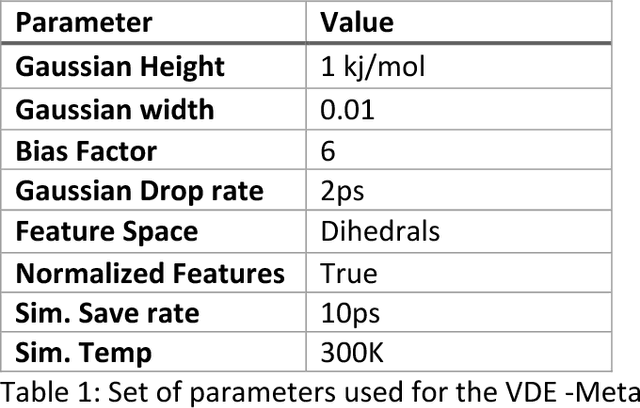
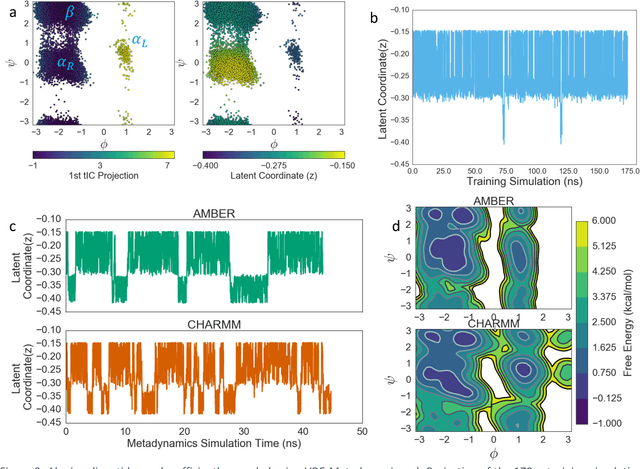
Variational auto-encoder frameworks have demonstrated success in reducing complex nonlinear dynamics in molecular simulation to a single non-linear embedding. In this work, we illustrate how this non-linear latent embedding can be used as a collective variable for enhanced sampling, and present a simple modification that allows us to rapidly perform sampling in multiple related systems. We first demonstrate our method is able to describe the effects of force field changes in capped alanine dipeptide after learning a model using AMBER99. We further provide a simple extension to variational dynamics encoders that allows the model to be trained in a more efficient manner on larger systems by encoding the outputs of a linear transformation using time-structure based independent component analysis (tICA). Using this technique, we show how such a model trained for one protein, the WW domain, can efficiently be transferred to perform enhanced sampling on a related mutant protein, the GTT mutation. This method shows promise for its ability to rapidly sample related systems using a single transferable collective variable and is generally applicable to sets of related simulations, enabling us to probe the effects of variation in increasingly large systems of biophysical interest.