 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAPEX-Agents
Jan 20, 2026We introduce the AI Productivity Index for Agents (APEX-Agents), a benchmark for assessing whether AI agents can execute long-horizon, cross-application tasks created by investment banking analysts, management consultants, and corporate lawyers. APEX-Agents requires agents to navigate realistic work environments with files and tools. We test eight agents for the leaderboard using Pass@1. Gemini 3 Flash (Thinking=High) achieves the highest score of 24.0%, followed by GPT-5.2 (Thinking=High), Claude Opus 4.5 (Thinking=High), and Gemini 3 Pro (Thinking=High). We open source the APEX-Agents benchmark (n=480) with all prompts, rubrics, gold outputs, files, and metadata. We also open-source Archipelago, our infrastructure for agent execution and evaluation.
PotentialNet for Molecular Property Prediction
Oct 22, 2018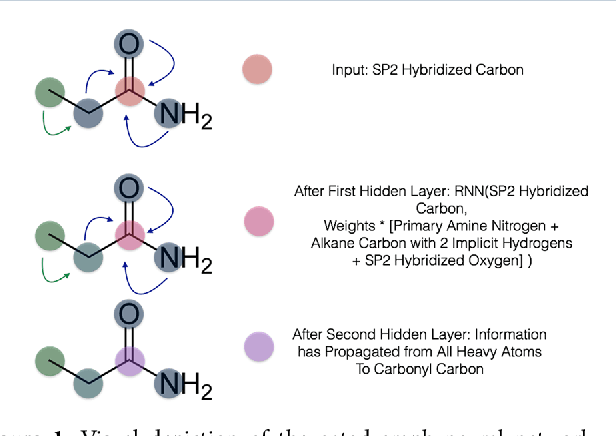
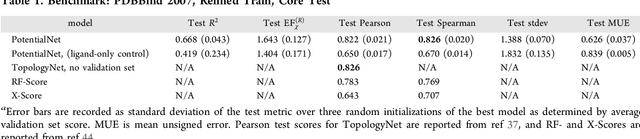
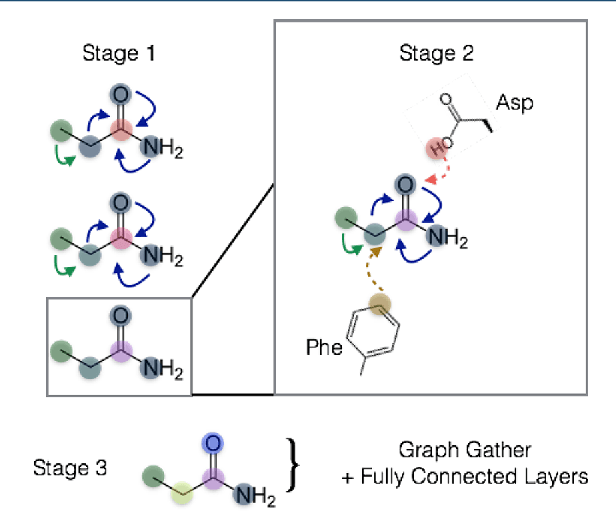
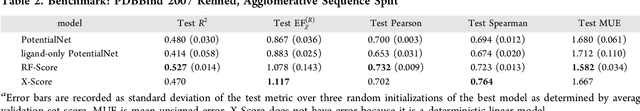
The arc of drug discovery entails a multiparameter optimization problem spanning vast length scales. They key parameters range from solubility (angstroms) to protein-ligand binding (nanometers) to in vivo toxicity (meters). Through feature learning---instead of feature engineering---deep neural networks promise to outperform both traditional physics-based and knowledge-based machine learning models for predicting molecular properties pertinent to drug discovery. To this end, we present the PotentialNet family of graph convolutions. These models are specifically designed for and achieve state-of-the-art performance for protein-ligand binding affinity. We further validate these deep neural networks by setting new standards of performance in several ligand-based tasks. In parallel, we introduce a new metric, the Regression Enrichment Factor $EF_\chi^{(R)}$, to measure the early enrichment of computational models for chemical data. Finally, we introduce a cross-validation strategy based on structural homology clustering that can more accurately measure model generalizability, which crucially distinguishes the aims of machine learning for drug discovery from standard machine learning tasks.