 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCompositional Deep Probabilistic Models of DNA Encoded Libraries
Oct 20, 2023DNA-Encoded Library (DEL) has proven to be a powerful tool that utilizes combinatorially constructed small molecules to facilitate highly-efficient screening assays. These selection experiments, involving multiple stages of washing, elution, and identification of potent binders via unique DNA barcodes, often generate complex data. This complexity can potentially mask the underlying signals, necessitating the application of computational tools such as machine learning to uncover valuable insights. We introduce a compositional deep probabilistic model of DEL data, DEL-Compose, which decomposes molecular representations into their mono-synthon, di-synthon, and tri-synthon building blocks and capitalizes on the inherent hierarchical structure of these molecules by modeling latent reactions between embedded synthons. Additionally, we investigate methods to improve the observation models for DEL count data such as integrating covariate factors to more effectively account for data noise. Across two popular public benchmark datasets (CA-IX and HRP), our model demonstrates strong performance compared to count baselines, enriches the correct pharmacophores, and offers valuable insights via its intrinsic interpretable structure, thereby providing a robust tool for the analysis of DEL data.
DEL-Dock: Molecular Docking-Enabled Modeling of DNA-Encoded Libraries
Dec 14, 2022DNA-Encoded Library (DEL) technology has enabled significant advances in hit identification by enabling efficient testing of combinatorially-generated molecular libraries. DEL screens measure protein binding affinity though sequencing reads of molecules tagged with unique DNA-barcodes that survive a series of selection experiments. Computational models have been deployed to learn the latent binding affinities that are correlated to the sequenced count data; however, this correlation is often obfuscated by various sources of noise introduced in its complicated data-generation process. In order to denoise DEL count data and screen for molecules with good binding affinity, computational models require the correct assumptions in their modeling structure to capture the correct signals underlying the data. Recent advances in DEL models have focused on probabilistic formulations of count data, but existing approaches have thus far been limited to only utilizing 2-D molecule-level representations. We introduce a new paradigm, DEL-Dock, that combines ligand-based descriptors with 3-D spatial information from docked protein-ligand complexes. 3-D spatial information allows our model to learn over the actual binding modality rather than using only structured-based information of the ligand. We show that our model is capable of effectively denoising DEL count data to predict molecule enrichment scores that are better correlated with experimental binding affinity measurements compared to prior works. Moreover, by learning over a collection of docked poses we demonstrate that our model, trained only on DEL data, implicitly learns to perform good docking pose selection without requiring external supervision from expensive-to-source protein crystal structures.
Automated design of collective variables using supervised machine learning
May 13, 2018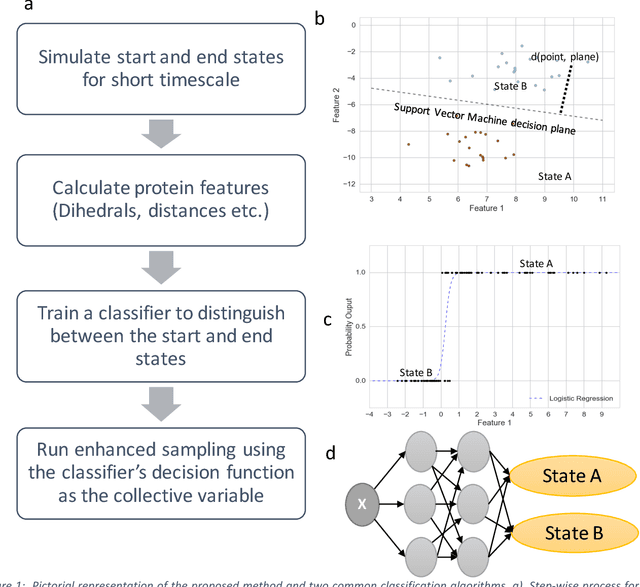
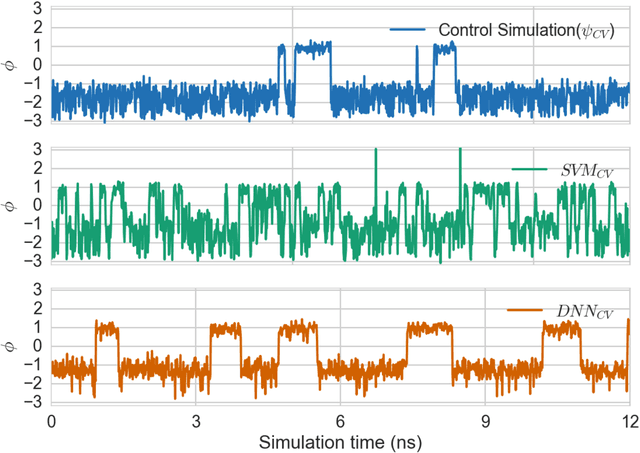
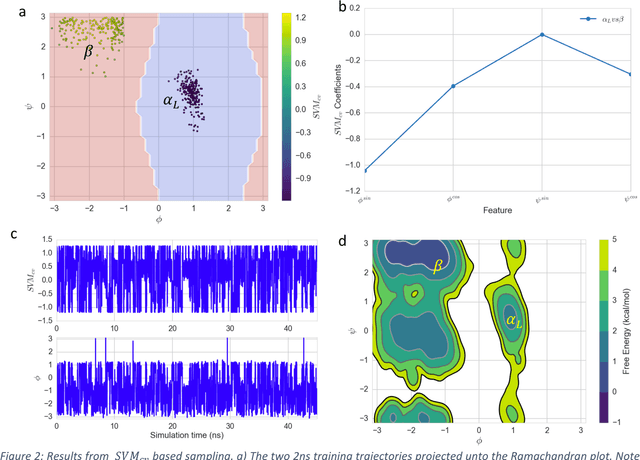

Selection of appropriate collective variables for enhancing sampling of molecular simulations remains an unsolved problem in computational biophysics. In particular, picking initial collective variables (CVs) is particularly challenging in higher dimensions. Which atomic coordinates or transforms there of from a list of thousands should one pick for enhanced sampling runs? How does a modeler even begin to pick starting coordinates for investigation? This remains true even in the case of simple two state systems and only increases in difficulty for multi-state systems. In this work, we solve the initial CV problem using a data-driven approach inspired by the filed of supervised machine learning. In particular, we show how the decision functions in supervised machine learning (SML) algorithms can be used as initial CVs (SML_cv) for accelerated sampling. Using solvated alanine dipeptide and Chignolin mini-protein as our test cases, we illustrate how the distance to the Support Vector Machines' decision hyperplane, the output probability estimates from Logistic Regression, the outputs from deep neural network classifiers, and other classifiers may be used to reversibly sample slow structural transitions. We discuss the utility of other SML algorithms that might be useful for identifying CVs for accelerating molecular simulations.
Using Deep Learning for Segmentation and Counting within Microscopy Data
Feb 28, 2018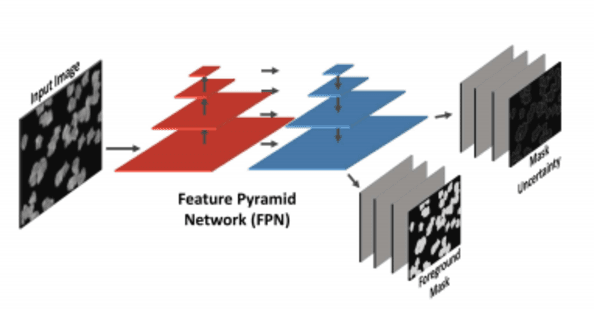
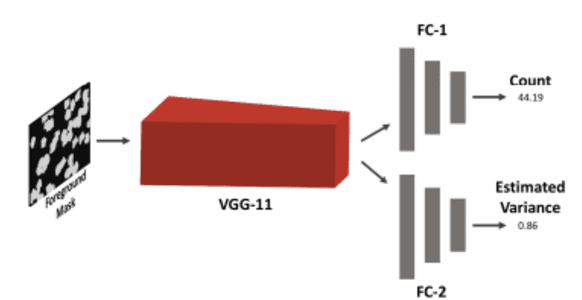
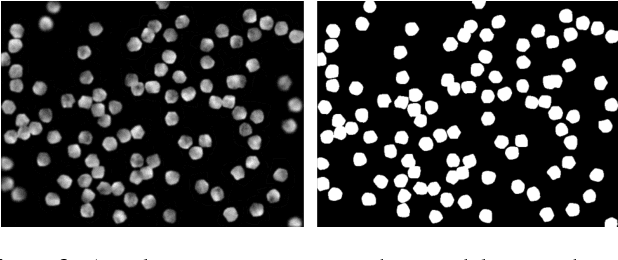

Cell counting is a ubiquitous, yet tedious task that would greatly benefit from automation. From basic biological questions to clinical trials, cell counts provide key quantitative feedback that drive research. Unfortunately, cell counting is most commonly a manual task and can be time-intensive. The task is made even more difficult due to overlapping cells, existence of multiple focal planes, and poor imaging quality, among other factors. Here, we describe a convolutional neural network approach, using a recently described feature pyramid network combined with a VGG-style neural network, for segmenting and subsequent counting of cells in a given microscopy image.
Transferable neural networks for enhanced sampling of protein dynamics
Jan 02, 2018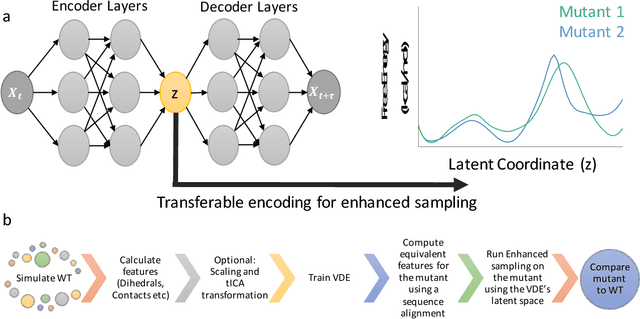
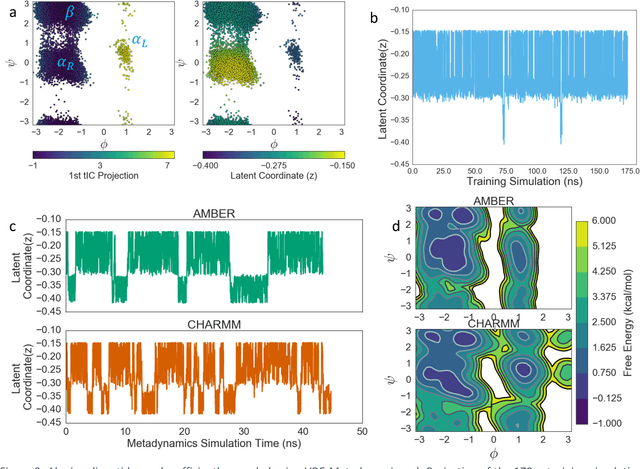
Variational auto-encoder frameworks have demonstrated success in reducing complex nonlinear dynamics in molecular simulation to a single non-linear embedding. In this work, we illustrate how this non-linear latent embedding can be used as a collective variable for enhanced sampling, and present a simple modification that allows us to rapidly perform sampling in multiple related systems. We first demonstrate our method is able to describe the effects of force field changes in capped alanine dipeptide after learning a model using AMBER99. We further provide a simple extension to variational dynamics encoders that allows the model to be trained in a more efficient manner on larger systems by encoding the outputs of a linear transformation using time-structure based independent component analysis (tICA). Using this technique, we show how such a model trained for one protein, the WW domain, can efficiently be transferred to perform enhanced sampling on a related mutant protein, the GTT mutation. This method shows promise for its ability to rapidly sample related systems using a single transferable collective variable and is generally applicable to sets of related simulations, enabling us to probe the effects of variation in increasingly large systems of biophysical interest.
Variational Encoding of Complex Dynamics
Dec 02, 2017
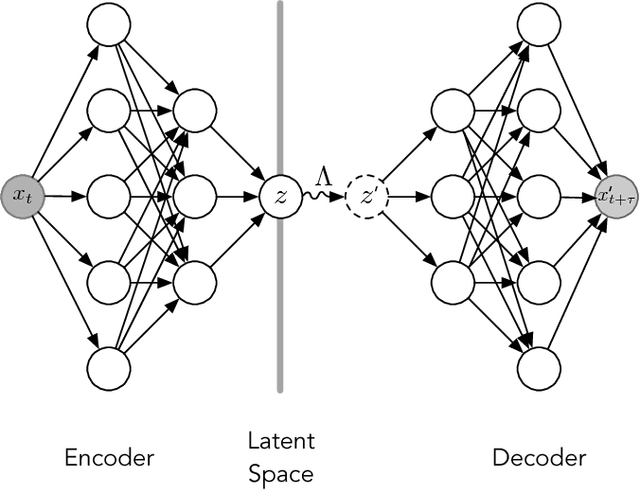
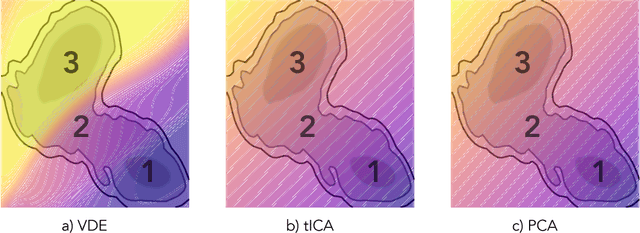
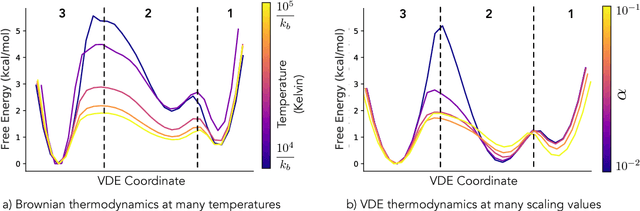
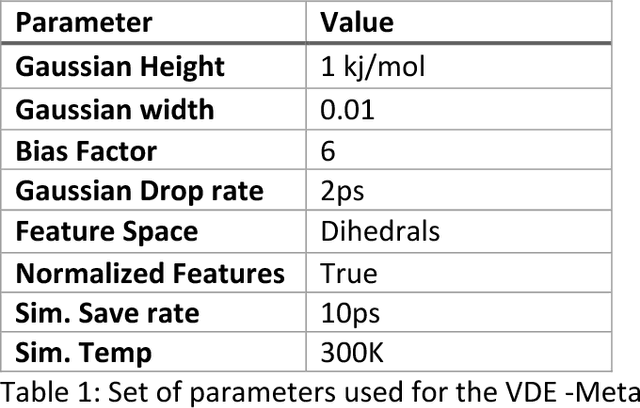
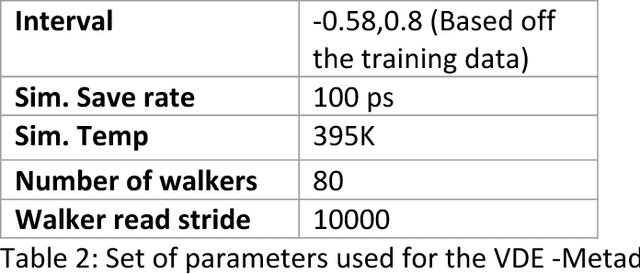
Often the analysis of time-dependent chemical and biophysical systems produces high-dimensional time-series data for which it can be difficult to interpret which individual features are most salient. While recent work from our group and others has demonstrated the utility of time-lagged co-variate models to study such systems, linearity assumptions can limit the compression of inherently nonlinear dynamics into just a few characteristic components. Recent work in the field of deep learning has led to the development of variational autoencoders (VAE), which are able to compress complex datasets into simpler manifolds. We present the use of a time-lagged VAE, or variational dynamics encoder (VDE), to reduce complex, nonlinear processes to a single embedding with high fidelity to the underlying dynamics. We demonstrate how the VDE is able to capture nontrivial dynamics in a variety of examples, including Brownian dynamics and atomistic protein folding. Additionally, we demonstrate a method for analyzing the VDE model, inspired by saliency mapping, to determine what features are selected by the VDE model to describe dynamics. The VDE presents an important step in applying techniques from deep learning to more accurately model and interpret complex biophysics.
* Fixed typos and added references
Understanding Protein Dynamics with L1-Regularized Reversible Hidden Markov Models
May 06, 2014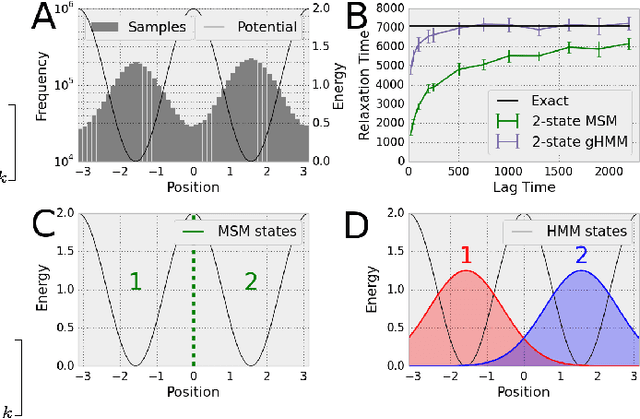
We present a machine learning framework for modeling protein dynamics. Our approach uses L1-regularized, reversible hidden Markov models to understand large protein datasets generated via molecular dynamics simulations. Our model is motivated by three design principles: (1) the requirement of massive scalability; (2) the need to adhere to relevant physical law; and (3) the necessity of providing accessible interpretations, critical for both cellular biology and rational drug design. We present an EM algorithm for learning and introduce a model selection criteria based on the physical notion of convergence in relaxation timescales. We contrast our model with standard methods in biophysics and demonstrate improved robustness. We implement our algorithm on GPUs and apply the method to two large protein simulation datasets generated respectively on the NCSA Bluewaters supercomputer and the Folding@Home distributed computing network. Our analysis identifies the conformational dynamics of the ubiquitin protein critical to cellular signaling, and elucidates the stepwise activation mechanism of the c-Src kinase protein.