 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeKinDEL: DNA-Encoded Library Dataset for Kinase Inhibitors
Oct 11, 2024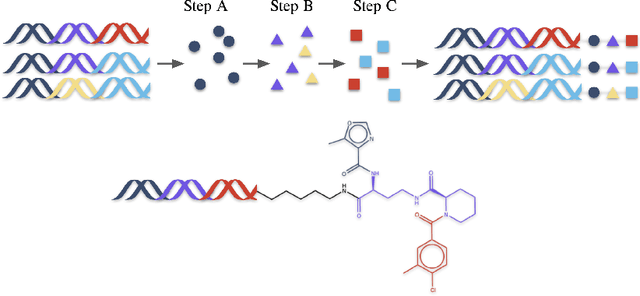

DNA-Encoded Libraries (DEL) are combinatorial small molecule libraries that offer an efficient way to characterize diverse chemical spaces. Selection experiments using DELs are pivotal to drug discovery efforts, enabling high-throughput screens for hit finding. However, limited availability of public DEL datasets hinders the advancement of computational techniques designed to process such data. To bridge this gap, we present KinDEL, one of the first large, publicly available DEL datasets on two kinases: Mitogen-Activated Protein Kinase 14 (MAPK14) and Discoidin Domain Receptor Tyrosine Kinase 1 (DDR1). Interest in this data modality is growing due to its ability to generate extensive supervised chemical data that densely samples around select molecular structures. Demonstrating one such application of the data, we benchmark different machine learning techniques to develop predictive models for hit identification; in particular, we highlight recent structure-based probabilistic approaches. Finally, we provide biophysical assay data, both on- and off-DNA, to validate our models on a smaller subset of molecules. Data and code for our benchmarks can be found at: https://github.com/insitro/kindel.
Compositional Deep Probabilistic Models of DNA Encoded Libraries
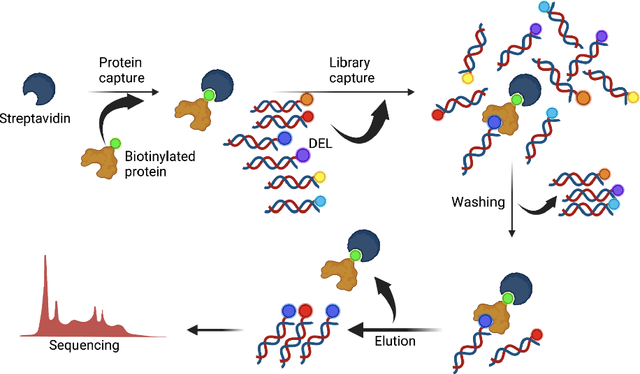
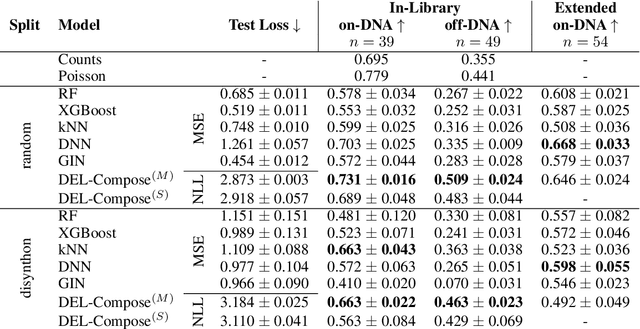
Oct 20, 2023DNA-Encoded Library (DEL) has proven to be a powerful tool that utilizes combinatorially constructed small molecules to facilitate highly-efficient screening assays. These selection experiments, involving multiple stages of washing, elution, and identification of potent binders via unique DNA barcodes, often generate complex data. This complexity can potentially mask the underlying signals, necessitating the application of computational tools such as machine learning to uncover valuable insights. We introduce a compositional deep probabilistic model of DEL data, DEL-Compose, which decomposes molecular representations into their mono-synthon, di-synthon, and tri-synthon building blocks and capitalizes on the inherent hierarchical structure of these molecules by modeling latent reactions between embedded synthons. Additionally, we investigate methods to improve the observation models for DEL count data such as integrating covariate factors to more effectively account for data noise. Across two popular public benchmark datasets (CA-IX and HRP), our model demonstrates strong performance compared to count baselines, enriches the correct pharmacophores, and offers valuable insights via its intrinsic interpretable structure, thereby providing a robust tool for the analysis of DEL data.
DEL-Dock: Molecular Docking-Enabled Modeling of DNA-Encoded Libraries
Dec 14, 2022DNA-Encoded Library (DEL) technology has enabled significant advances in hit identification by enabling efficient testing of combinatorially-generated molecular libraries. DEL screens measure protein binding affinity though sequencing reads of molecules tagged with unique DNA-barcodes that survive a series of selection experiments. Computational models have been deployed to learn the latent binding affinities that are correlated to the sequenced count data; however, this correlation is often obfuscated by various sources of noise introduced in its complicated data-generation process. In order to denoise DEL count data and screen for molecules with good binding affinity, computational models require the correct assumptions in their modeling structure to capture the correct signals underlying the data. Recent advances in DEL models have focused on probabilistic formulations of count data, but existing approaches have thus far been limited to only utilizing 2-D molecule-level representations. We introduce a new paradigm, DEL-Dock, that combines ligand-based descriptors with 3-D spatial information from docked protein-ligand complexes. 3-D spatial information allows our model to learn over the actual binding modality rather than using only structured-based information of the ligand. We show that our model is capable of effectively denoising DEL count data to predict molecule enrichment scores that are better correlated with experimental binding affinity measurements compared to prior works. Moreover, by learning over a collection of docked poses we demonstrate that our model, trained only on DEL data, implicitly learns to perform good docking pose selection without requiring external supervision from expensive-to-source protein crystal structures.
Fragment-based Sequential Translation for Molecular Optimization
Oct 26, 2021



Searching for novel molecular compounds with desired properties is an important problem in drug discovery. Many existing frameworks generate molecules one atom at a time. We instead propose a flexible editing paradigm that generates molecules using learned molecular fragments--meaningful substructures of molecules. To do so, we train a variational autoencoder (VAE) to encode molecular fragments in a coherent latent space, which we then utilize as a vocabulary for editing molecules to explore the complex chemical property space. Equipped with the learned fragment vocabulary, we propose Fragment-based Sequential Translation (FaST), which learns a reinforcement learning (RL) policy to iteratively translate model-discovered molecules into increasingly novel molecules while satisfying desired properties. Empirical evaluation shows that FaST significantly improves over state-of-the-art methods on benchmark single/multi-objective molecular optimization tasks.
Optimal Transport Graph Neural Networks
Jun 11, 2020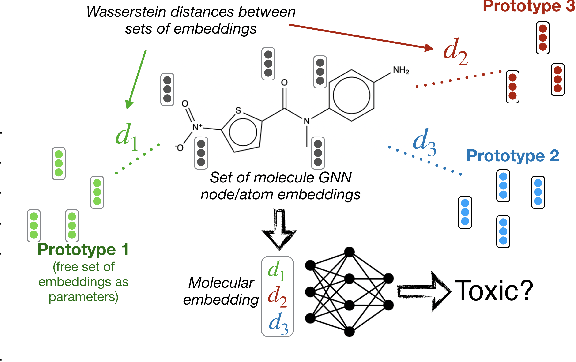
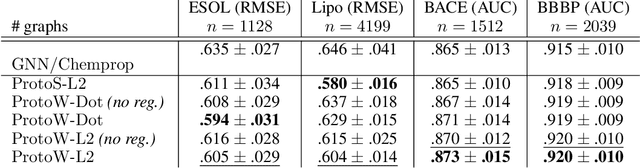
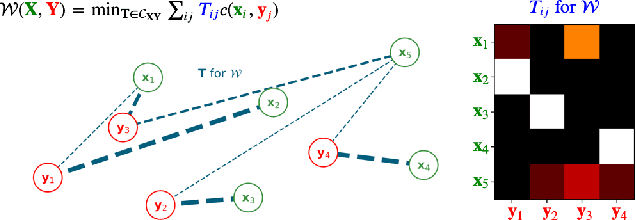
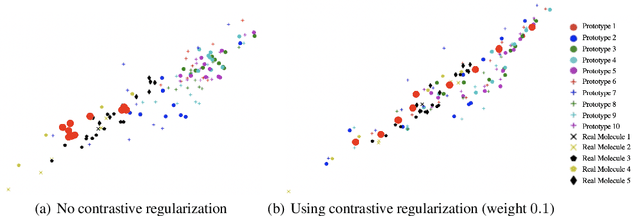
Current graph neural network (GNN) architectures naively average or sum node embeddings into an aggregated graph representation---potentially losing structural or semantic information. We here introduce OT-GNN that compute graph embeddings from optimal transport distances between the set of GNN node embeddings and "prototype" point clouds as free parameters. This allows different prototypes to highlight key facets of different graph subparts. We show that our function class on point clouds satisfies a universal approximation theorem, a fundamental property which was lost by sum aggregation. Nevertheless, empirically the model has a natural tendency to collapse back to the standard aggregation during training. We address this optimization issue by proposing an efficient noise contrastive regularizer, steering the model towards truly exploiting the optimal transport geometry. Our model consistently exhibits better generalization performance on several molecular property prediction tasks, yielding also smoother representations.
Learning to Make Generalizable and Diverse Predictions for Retrosynthesis
Oct 21, 2019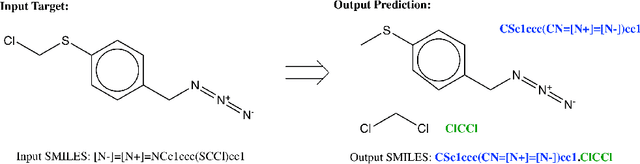
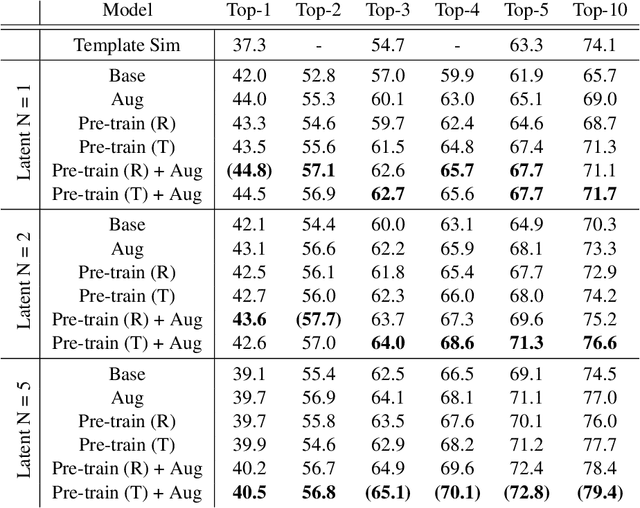
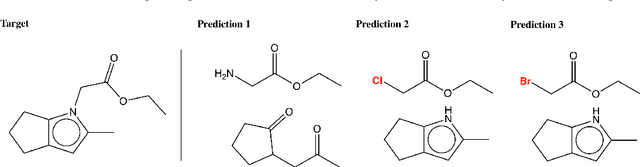
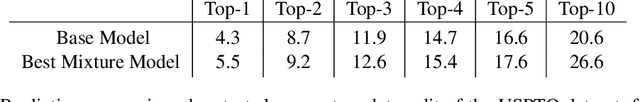
We propose a new model for making generalizable and diverse retrosynthetic reaction predictions. Given a target compound, the task is to predict the likely chemical reactants to produce the target. This generative task can be framed as a sequence-to-sequence problem by using the SMILES representations of the molecules. Building on top of the popular Transformer architecture, we propose two novel pre-training methods that construct relevant auxiliary tasks (plausible reactions) for our problem. Furthermore, we incorporate a discrete latent variable model into the architecture to encourage the model to produce a diverse set of alternative predictions. On the 50k subset of reaction examples from the United States patent literature (USPTO-50k) benchmark dataset, our model greatly improves performance over the baseline, while also generating predictions that are more diverse.
Path-Augmented Graph Transformer Network
May 29, 2019
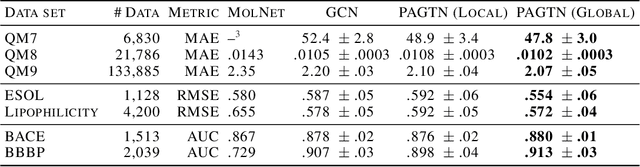


Much of the recent work on learning molecular representations has been based on Graph Convolution Networks (GCN). These models rely on local aggregation operations and can therefore miss higher-order graph properties. To remedy this, we propose Path-Augmented Graph Transformer Networks (PAGTN) that are explicitly built on longer-range dependencies in graph-structured data. Specifically, we use path features in molecular graphs to create global attention layers. We compare our PAGTN model against the GCN model and show that our model consistently outperforms GCNs on molecular property prediction datasets including quantum chemistry (QM7, QM8, QM9), physical chemistry (ESOL, Lipophilictiy) and biochemistry (BACE, BBBP).