 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFragment-Wise Interpretability in Graph Neural Networks via Molecule Decomposition and Contribution Analysis
Aug 20, 2025Graph neural networks have demonstrated remarkable success in predicting molecular properties by leveraging the rich structural information encoded in molecular graphs. However, their black-box nature reduces interpretability, which limits trust in their predictions for important applications such as drug discovery and materials design. Furthermore, existing explanation techniques often fail to reliably quantify the contribution of individual atoms or substructures due to the entangled message-passing dynamics. We introduce SEAL (Substructure Explanation via Attribution Learning), a new interpretable graph neural network that attributes model predictions to meaningful molecular subgraphs. SEAL decomposes input graphs into chemically relevant fragments and estimates their causal influence on the output. The strong alignment between fragment contributions and model predictions is achieved by explicitly reducing inter-fragment message passing in our proposed model architecture. Extensive evaluations on synthetic benchmarks and real-world molecular datasets demonstrate that SEAL outperforms other explainability methods in both quantitative attribution metrics and human-aligned interpretability. A user study further confirms that SEAL provides more intuitive and trustworthy explanations to domain experts. By bridging the gap between predictive performance and interpretability, SEAL offers a promising direction for more transparent and actionable molecular modeling.
B-XAIC Dataset: Benchmarking Explainable AI for Graph Neural Networks Using Chemical Data
May 28, 2025Understanding the reasoning behind deep learning model predictions is crucial in cheminformatics and drug discovery, where molecular design determines their properties. However, current evaluation frameworks for Explainable AI (XAI) in this domain often rely on artificial datasets or simplified tasks, employing data-derived metrics that fail to capture the complexity of real-world scenarios and lack a direct link to explanation faithfulness. To address this, we introduce B-XAIC, a novel benchmark constructed from real-world molecular data and diverse tasks with known ground-truth rationales for assigned labels. Through a comprehensive evaluation using B-XAIC, we reveal limitations of existing XAI methods for Graph Neural Networks (GNNs) in the molecular domain. This benchmark provides a valuable resource for gaining deeper insights into the faithfulness of XAI, facilitating the development of more reliable and interpretable models.
KinDEL: DNA-Encoded Library Dataset for Kinase Inhibitors
Oct 11, 2024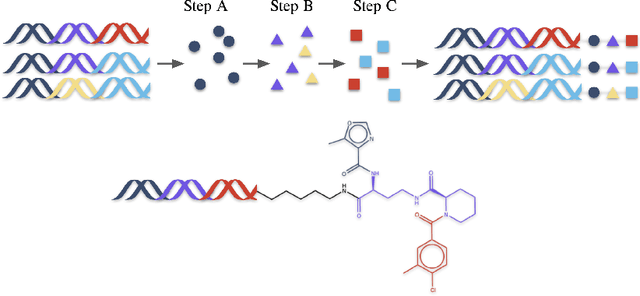

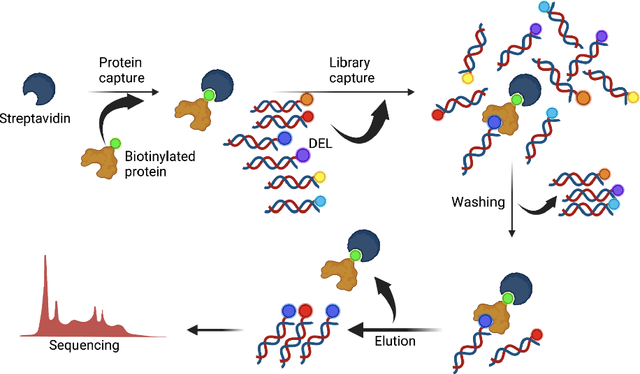
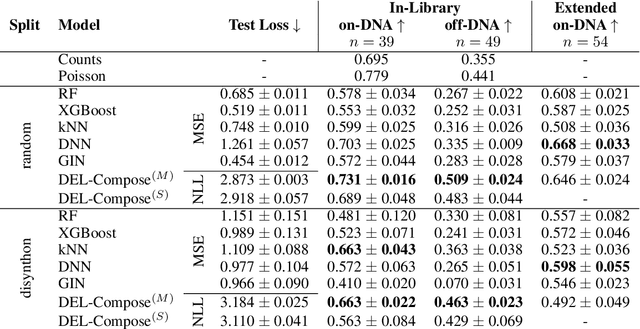
DNA-Encoded Libraries (DEL) are combinatorial small molecule libraries that offer an efficient way to characterize diverse chemical spaces. Selection experiments using DELs are pivotal to drug discovery efforts, enabling high-throughput screens for hit finding. However, limited availability of public DEL datasets hinders the advancement of computational techniques designed to process such data. To bridge this gap, we present KinDEL, one of the first large, publicly available DEL datasets on two kinases: Mitogen-Activated Protein Kinase 14 (MAPK14) and Discoidin Domain Receptor Tyrosine Kinase 1 (DDR1). Interest in this data modality is growing due to its ability to generate extensive supervised chemical data that densely samples around select molecular structures. Demonstrating one such application of the data, we benchmark different machine learning techniques to develop predictive models for hit identification; in particular, we highlight recent structure-based probabilistic approaches. Finally, we provide biophysical assay data, both on- and off-DNA, to validate our models on a smaller subset of molecules. Data and code for our benchmarks can be found at: https://github.com/insitro/kindel.
ProGReST: Prototypical Graph Regression Soft Trees for Molecular Property Prediction
Oct 07, 2022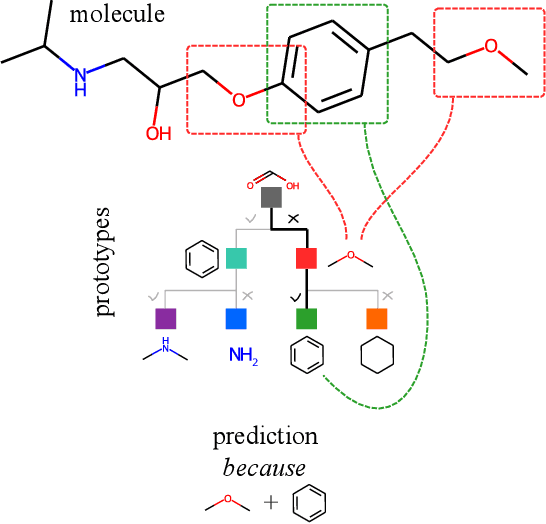
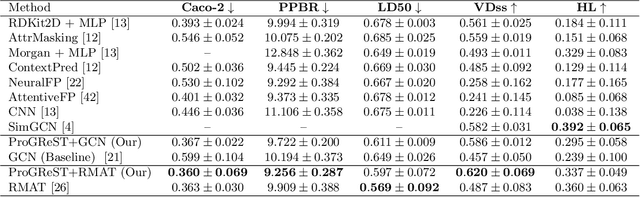
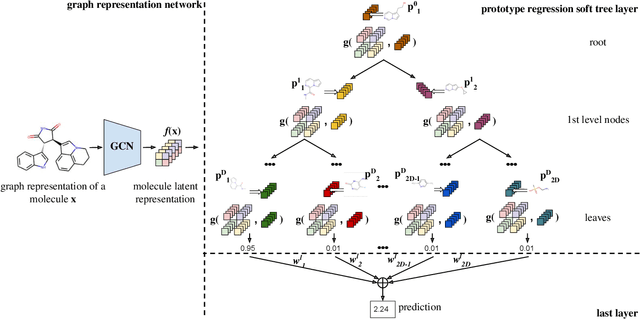

In this work, we propose the novel Prototypical Graph Regression Self-explainable Trees (ProGReST) model, which combines prototype learning, soft decision trees, and Graph Neural Networks. In contrast to other works, our model can be used to address various challenging tasks, including compound property prediction. In ProGReST, the rationale is obtained along with prediction due to the model's built-in interpretability. Additionally, we introduce a new graph prototype projection to accelerate model training. Finally, we evaluate PRoGReST on a wide range of chemical datasets for molecular property prediction and perform in-depth analysis with chemical experts to evaluate obtained interpretations. Our method achieves competitive results against state-of-the-art methods.
Relative Molecule Self-Attention Transformer
Oct 12, 2021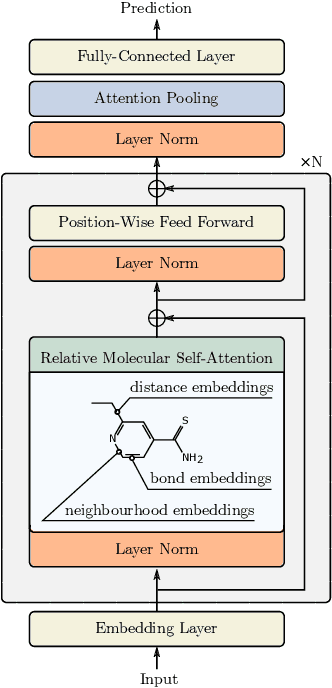
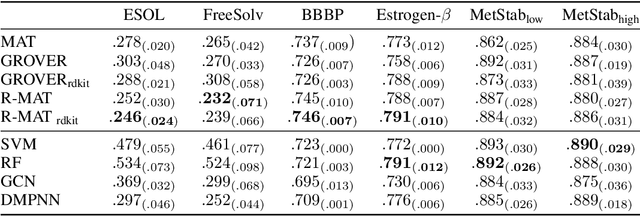
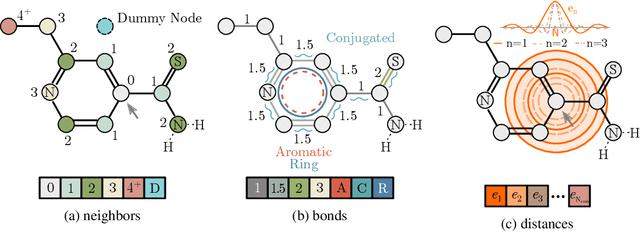
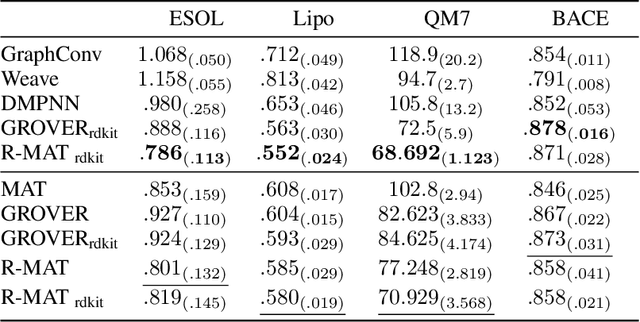
Self-supervised learning holds promise to revolutionize molecule property prediction - a central task to drug discovery and many more industries - by enabling data efficient learning from scarce experimental data. Despite significant progress, non-pretrained methods can be still competitive in certain settings. We reason that architecture might be a key bottleneck. In particular, enriching the backbone architecture with domain-specific inductive biases has been key for the success of self-supervised learning in other domains. In this spirit, we methodologically explore the design space of the self-attention mechanism tailored to molecular data. We identify a novel variant of self-attention adapted to processing molecules, inspired by the relative self-attention layer, which involves fusing embedded graph and distance relationships between atoms. Our main contribution is Relative Molecule Attention Transformer (R-MAT): a novel Transformer-based model based on the developed self-attention layer that achieves state-of-the-art or very competitive results across a~wide range of molecule property prediction tasks.
SONG: Self-Organizing Neural Graphs
Jul 28, 2021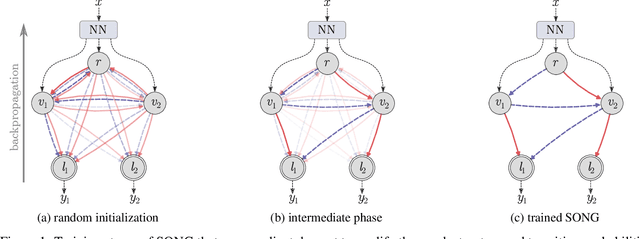



Recent years have seen a surge in research on deep interpretable neural networks with decision trees as one of the most commonly incorporated tools. There are at least three advantages of using decision trees over logistic regression classification models: they are easy to interpret since they are based on binary decisions, they can make decisions faster, and they provide a hierarchy of classes. However, one of the well-known drawbacks of decision trees, as compared to decision graphs, is that decision trees cannot reuse the decision nodes. Nevertheless, decision graphs were not commonly used in deep learning due to the lack of efficient gradient-based training techniques. In this paper, we fill this gap and provide a general paradigm based on Markov processes, which allows for efficient training of the special type of decision graphs, which we call Self-Organizing Neural Graphs (SONG). We provide an extensive theoretical study of SONG, complemented by experiments conducted on Letter, Connect4, MNIST, CIFAR, and TinyImageNet datasets, showing that our method performs on par or better than existing decision models.
Comparison of Atom Representations in Graph Neural Networks for Molecular Property Prediction
Nov 23, 2020

Graph neural networks have recently become a standard method for analysing chemical compounds. In the field of molecular property prediction, the emphasis is now put on designing new model architectures, and the importance of atom featurisation is oftentimes belittled. When contrasting two graph neural networks, the use of different atom features possibly leads to the incorrect attribution of the results to the network architecture. To provide a better understanding of this issue, we compare multiple atom representations for graph models and evaluate them on the prediction of free energy, solubility, and metabolic stability. To the best of our knowledge, this is the first methodological study that focuses on the relevance of atom representation to the predictive performance of graph neural networks.
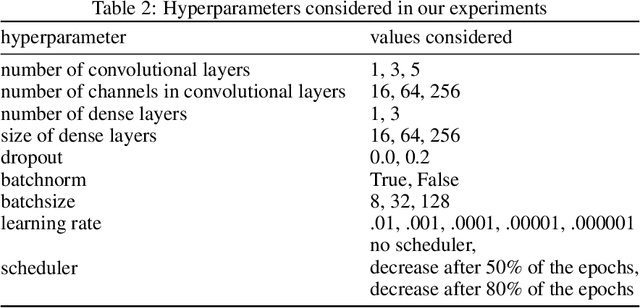
Processing of incomplete images by (graph) convolutional neural networks
Oct 26, 2020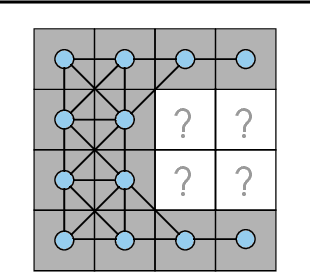
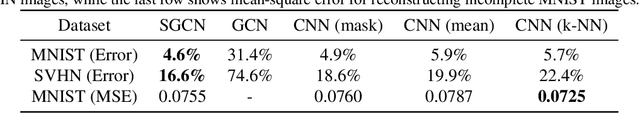
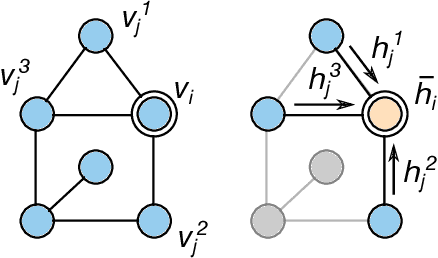

We investigate the problem of training neural networks from incomplete images without replacing missing values. For this purpose, we first represent an image as a graph, in which missing pixels are entirely ignored. The graph image representation is processed using a spatial graph convolutional network (SGCN) -- a type of graph convolutional networks, which is a proper generalization of classical CNNs operating on images. On one hand, our approach avoids the problem of missing data imputation while, on the other hand, there is a natural correspondence between CNNs and SGCN. Experiments confirm that our approach performs better than analogical CNNs with the imputation of missing values on typical classification and reconstruction tasks.
We Should at Least Be Able to Design Molecules That Dock Well
Jul 01, 2020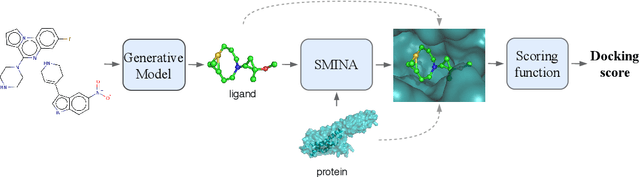
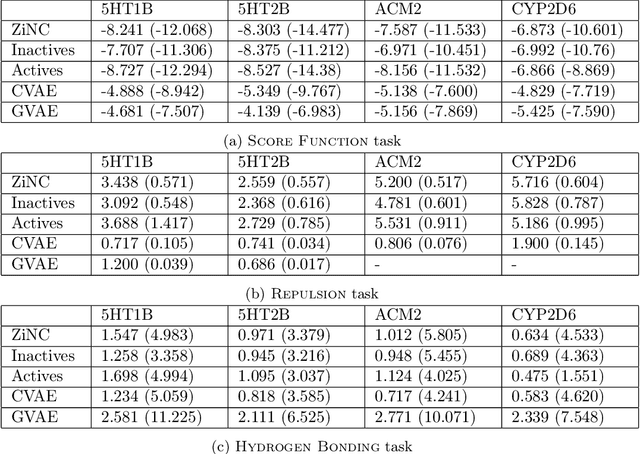
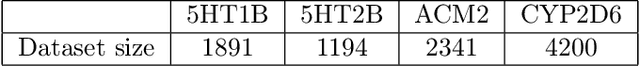
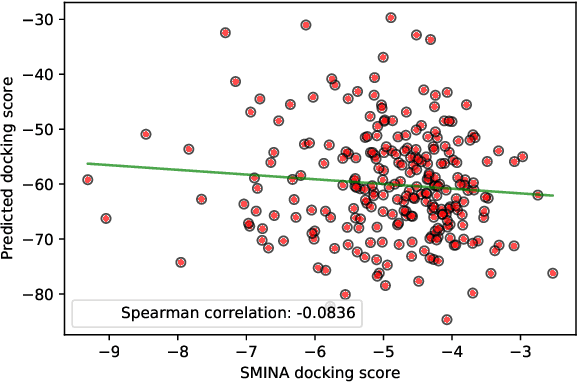
Designing compounds with desired properties is a key element of the drug discovery process. However, measuring progress in the field has been challenging due to the lack of realistic retrospective benchmarks, and the large cost of prospective validation. To close this gap, we propose a benchmark based on docking, a popular computational method for assessing molecule binding to a protein. Concretely, the goal is to generate drug-like molecules that are scored highly by SMINA, a popular docking software. We observe that popular graph-based generative models fail to generate molecules with a high docking score when trained using a realistically sized training set. This suggests a limitation of the current incarnation of models for de novo drug design. Finally, we propose a simplified version of the benchmark based on a simpler scoring function, and show that the tested models are able to partially solve it. We release the benchmark as an easy to use package available at https://github.com/cieplinski-tobiasz/smina-docking-benchmark. We hope that our benchmark will serve as a stepping stone towards the goal of automatically generating promising drug candidates.
Molecule Attention Transformer
Feb 19, 2020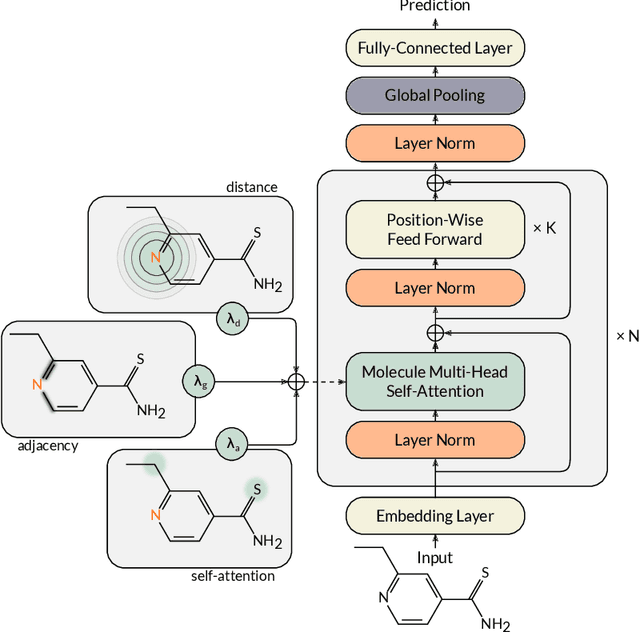

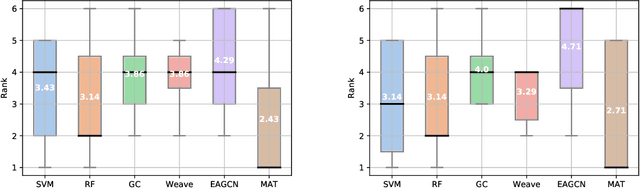
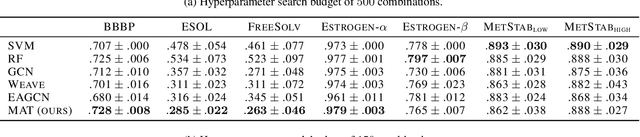
Designing a single neural network architecture that performs competitively across a range of molecule property prediction tasks remains largely an open challenge, and its solution may unlock a widespread use of deep learning in the drug discovery industry. To move towards this goal, we propose Molecule Attention Transformer (MAT). Our key innovation is to augment the attention mechanism in Transformer using inter-atomic distances and the molecular graph structure. Experiments show that MAT performs competitively on a diverse set of molecular prediction tasks. Most importantly, with a simple self-supervised pretraining, MAT requires tuning of only a few hyperparameter values to achieve state-of-the-art performance on downstream tasks. Finally, we show that attention weights learned by MAT are interpretable from the chemical point of view.