 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTSPP: A Unified Benchmarking Tool for Time-series Forecasting
Jan 08, 2024While machine learning has witnessed significant advancements, the emphasis has largely been on data acquisition and model creation. However, achieving a comprehensive assessment of machine learning solutions in real-world settings necessitates standardization throughout the entire pipeline. This need is particularly acute in time series forecasting, where diverse settings impede meaningful comparisons between various methods. To bridge this gap, we propose a unified benchmarking framework that exposes the crucial modelling and machine learning decisions involved in developing time series forecasting models. This framework fosters seamless integration of models and datasets, aiding both practitioners and researchers in their development efforts. We benchmark recently proposed models within this framework, demonstrating that carefully implemented deep learning models with minimal effort can rival gradient-boosting decision trees requiring extensive feature engineering and expert knowledge.
A Framework for Large Scale Synthetic Graph Dataset Generation
Oct 06, 2022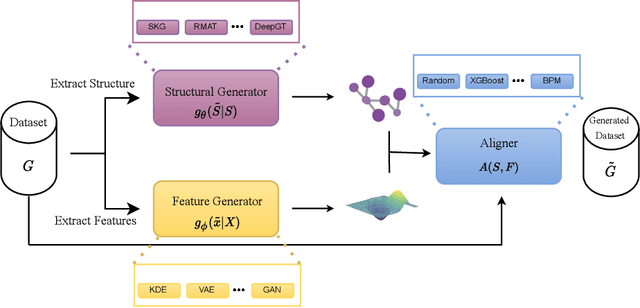
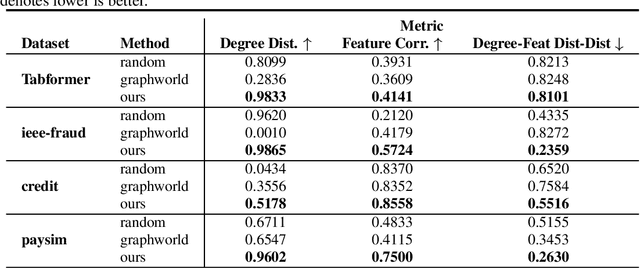
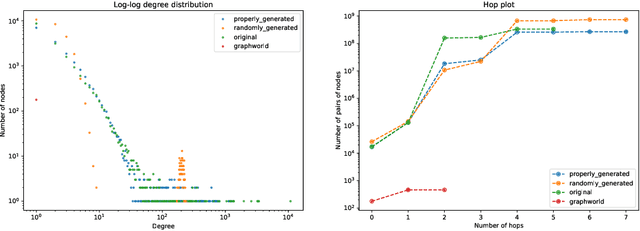
Recently there has been increasing interest in developing and deploying deep graph learning algorithms for many graph analysis tasks such as node and edge classification, link prediction, and clustering with numerous practical applications such as fraud detection, drug discovery, or recommender systems. Allbeit there is a limited number of publicly available graph-structured datasets, most of which are tiny compared to production-sized applications with trillions of edges and billions of nodes. Further, new algorithms and models are benchmarked across similar datasets with similar properties. In this work, we tackle this shortcoming by proposing a scalable synthetic graph generation tool that can mimic the original data distribution of real-world graphs and scale them to arbitrary sizes. This tool can be used then to learn a set of parametric models from proprietary datasets that can subsequently be released to researchers to study various graph methods on the synthetic data increasing prototype development and novel applications. Finally, the performance of the graph learning algorithms depends not only on the size but also on the dataset's structure. We show how our framework generalizes across a set of datasets, mimicking both structural and feature distributions as well as its scalability across varying dataset sizes.
Relative Molecule Self-Attention Transformer
Oct 12, 2021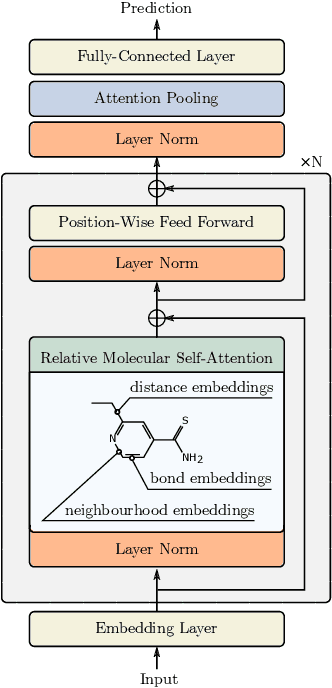
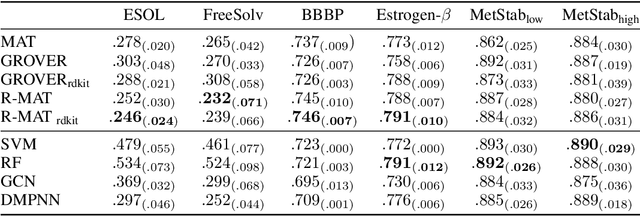
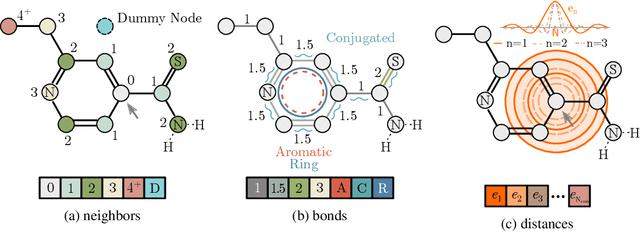
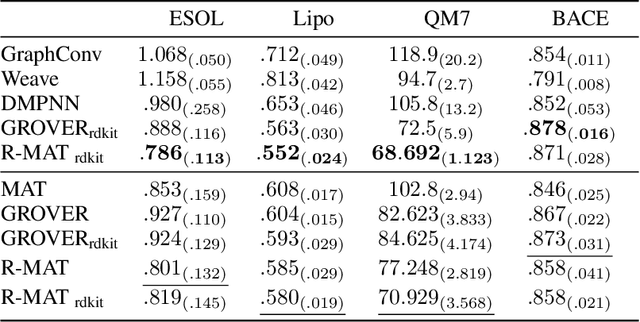
Self-supervised learning holds promise to revolutionize molecule property prediction - a central task to drug discovery and many more industries - by enabling data efficient learning from scarce experimental data. Despite significant progress, non-pretrained methods can be still competitive in certain settings. We reason that architecture might be a key bottleneck. In particular, enriching the backbone architecture with domain-specific inductive biases has been key for the success of self-supervised learning in other domains. In this spirit, we methodologically explore the design space of the self-attention mechanism tailored to molecular data. We identify a novel variant of self-attention adapted to processing molecules, inspired by the relative self-attention layer, which involves fusing embedded graph and distance relationships between atoms. Our main contribution is Relative Molecule Attention Transformer (R-MAT): a novel Transformer-based model based on the developed self-attention layer that achieves state-of-the-art or very competitive results across a~wide range of molecule property prediction tasks.