 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRelative Molecule Self-Attention Transformer
Paper and Code
Oct 12, 2021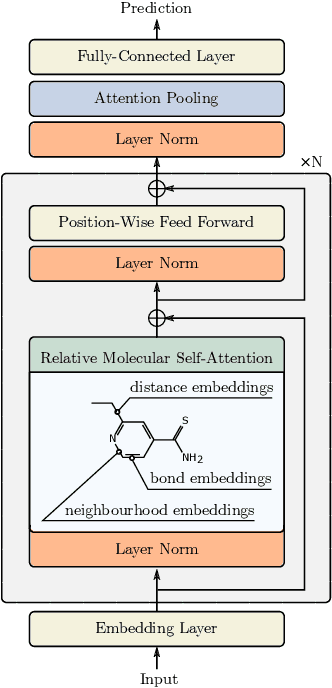
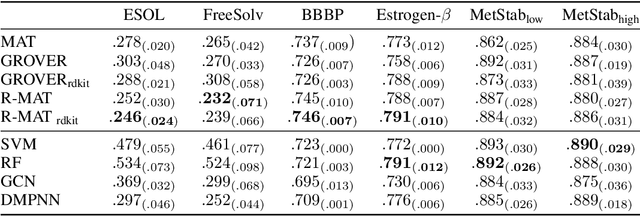
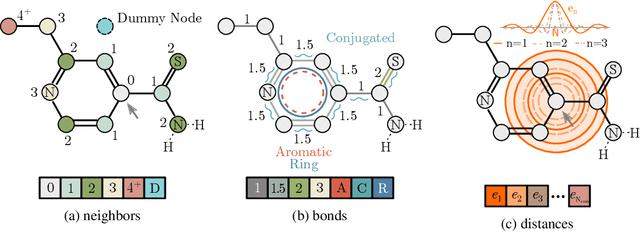
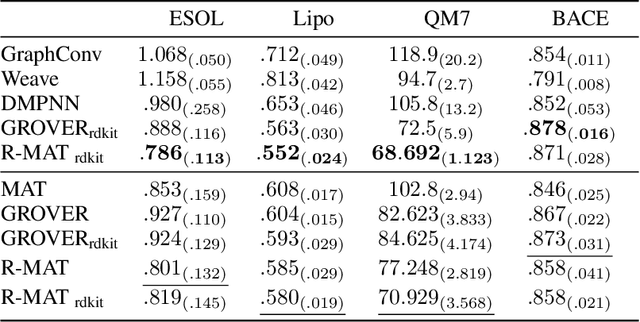
Self-supervised learning holds promise to revolutionize molecule property prediction - a central task to drug discovery and many more industries - by enabling data efficient learning from scarce experimental data. Despite significant progress, non-pretrained methods can be still competitive in certain settings. We reason that architecture might be a key bottleneck. In particular, enriching the backbone architecture with domain-specific inductive biases has been key for the success of self-supervised learning in other domains. In this spirit, we methodologically explore the design space of the self-attention mechanism tailored to molecular data. We identify a novel variant of self-attention adapted to processing molecules, inspired by the relative self-attention layer, which involves fusing embedded graph and distance relationships between atoms. Our main contribution is Relative Molecule Attention Transformer (R-MAT): a novel Transformer-based model based on the developed self-attention layer that achieves state-of-the-art or very competitive results across a~wide range of molecule property prediction tasks.