 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgetsGT: Stochastic Time Series Modeling With Transformer
Mar 15, 2024Time series methods are of fundamental importance in virtually any field of science that deals with temporally structured data. Recently, there has been a surge of deterministic transformer models with time series-specific architectural biases. In this paper, we go in a different direction by introducing tsGT, a stochastic time series model built on a general-purpose transformer architecture. We focus on using a well-known and theoretically justified rolling window backtesting and evaluation protocol. We show that tsGT outperforms the state-of-the-art models on MAD and RMSE, and surpasses its stochastic peers on QL and CRPS, on four commonly used datasets. We complement these results with a detailed analysis of tsGT's ability to model the data distribution and predict marginal quantile values.
Relative Molecule Self-Attention Transformer
Oct 12, 2021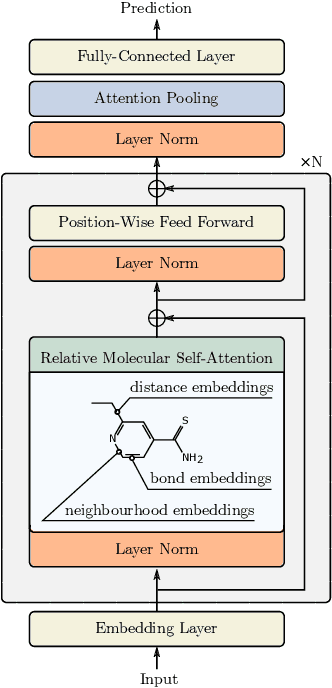
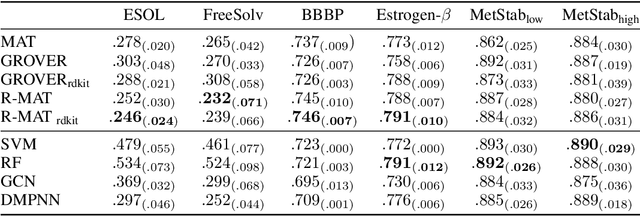
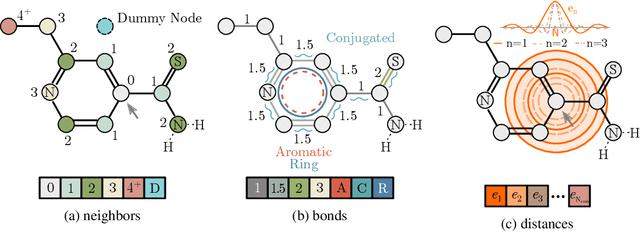
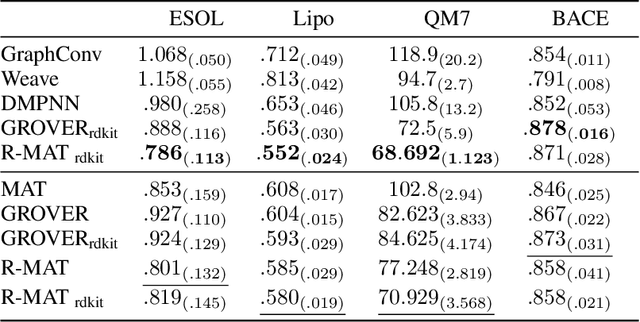
Self-supervised learning holds promise to revolutionize molecule property prediction - a central task to drug discovery and many more industries - by enabling data efficient learning from scarce experimental data. Despite significant progress, non-pretrained methods can be still competitive in certain settings. We reason that architecture might be a key bottleneck. In particular, enriching the backbone architecture with domain-specific inductive biases has been key for the success of self-supervised learning in other domains. In this spirit, we methodologically explore the design space of the self-attention mechanism tailored to molecular data. We identify a novel variant of self-attention adapted to processing molecules, inspired by the relative self-attention layer, which involves fusing embedded graph and distance relationships between atoms. Our main contribution is Relative Molecule Attention Transformer (R-MAT): a novel Transformer-based model based on the developed self-attention layer that achieves state-of-the-art or very competitive results across a~wide range of molecule property prediction tasks.
On the relationship between disentanglement and multi-task learning
Oct 07, 2021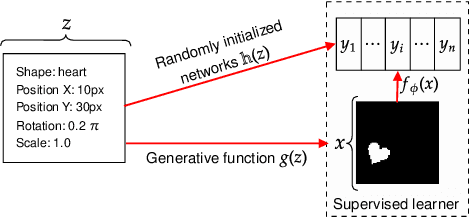

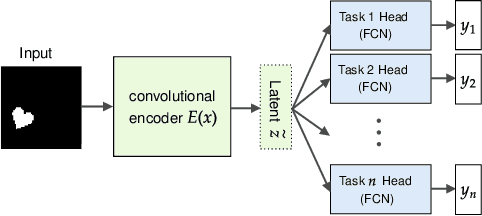
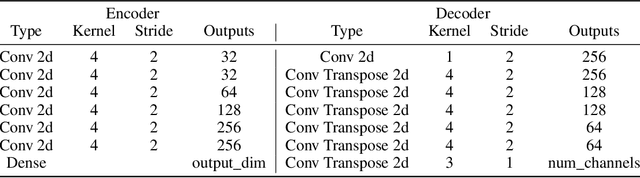
One of the main arguments behind studying disentangled representations is the assumption that they can be easily reused in different tasks. At the same time finding a joint, adaptable representation of data is one of the key challenges in the multi-task learning setting. In this paper, we take a closer look at the relationship between disentanglement and multi-task learning based on hard parameter sharing. We perform a thorough empirical study of the representations obtained by neural networks trained on automatically generated supervised tasks. Using a set of standard metrics we show that disentanglement appears naturally during the process of multi-task neural network training.
PluGeN: Multi-Label Conditional Generation From Pre-Trained Models
Sep 18, 2021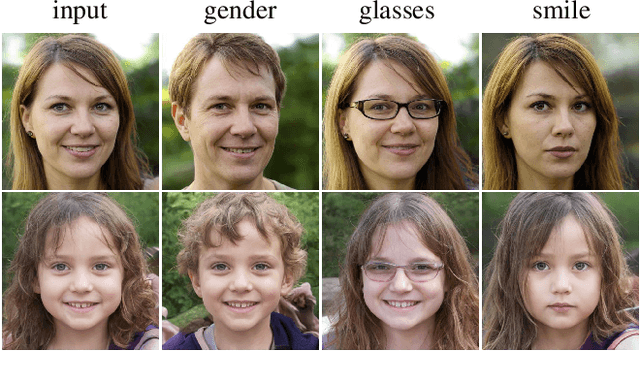
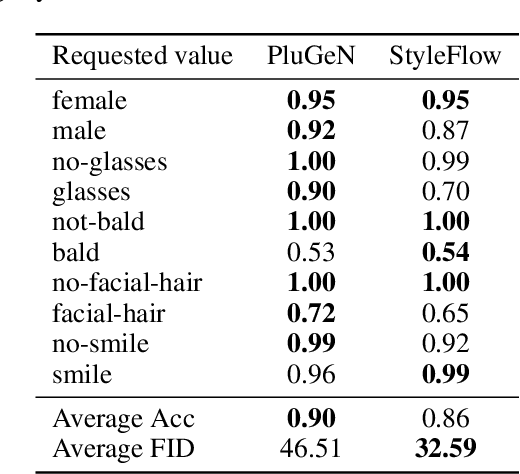
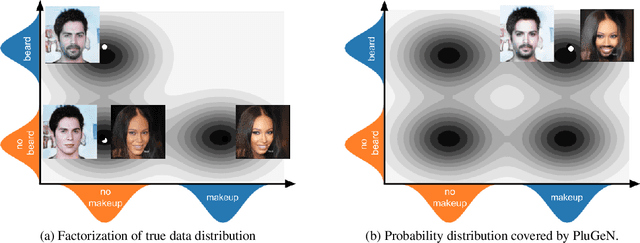
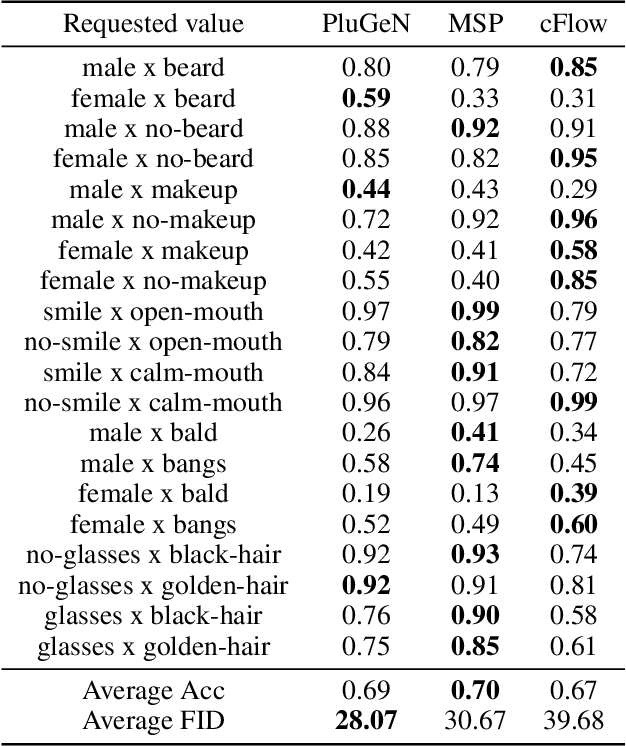
Modern generative models achieve excellent quality in a variety of tasks including image or text generation and chemical molecule modeling. However, existing methods often lack the essential ability to generate examples with requested properties, such as the age of the person in the photo or the weight of the generated molecule. Incorporating such additional conditioning factors would require rebuilding the entire architecture and optimizing the parameters from scratch. Moreover, it is difficult to disentangle selected attributes so that to perform edits of only one attribute while leaving the others unchanged. To overcome these limitations we propose PluGeN (Plugin Generative Network), a simple yet effective generative technique that can be used as a plugin to pre-trained generative models. The idea behind our approach is to transform the entangled latent representation using a flow-based module into a multi-dimensional space where the values of each attribute are modeled as an independent one-dimensional distribution. In consequence, PluGeN can generate new samples with desired attributes as well as manipulate labeled attributes of existing examples. Due to the disentangling of the latent representation, we are even able to generate samples with rare or unseen combinations of attributes in the dataset, such as a young person with gray hair, men with make-up, or women with beards. We combined PluGeN with GAN and VAE models and applied it to conditional generation and manipulation of images and chemical molecule modeling. Experiments demonstrate that PluGeN preserves the quality of backbone models while adding the ability to control the values of labeled attributes.
Flow-based SVDD for anomaly detection
Aug 10, 2021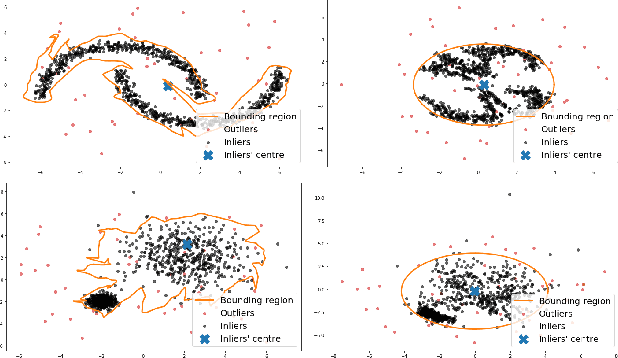
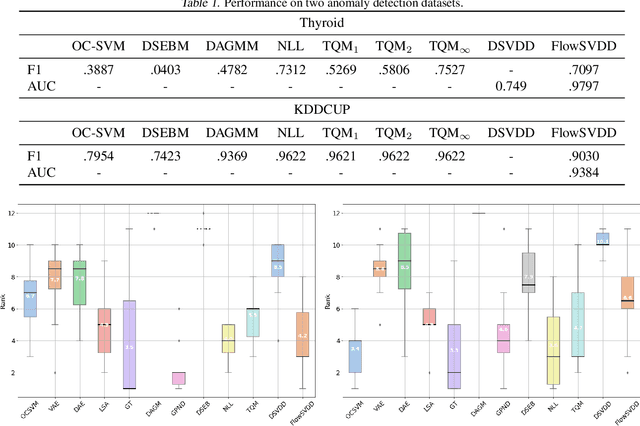

We propose FlowSVDD -- a flow-based one-class classifier for anomaly/outliers detection that realizes a well-known SVDD principle using deep learning tools. Contrary to other approaches to deep SVDD, the proposed model is instantiated using flow-based models, which naturally prevents from collapsing of bounding hypersphere into a single point. Experiments show that FlowSVDD achieves comparable results to the current state-of-the-art methods and significantly outperforms related deep SVDD methods on benchmark datasets.
Comparison of Atom Representations in Graph Neural Networks for Molecular Property Prediction
Nov 23, 2020

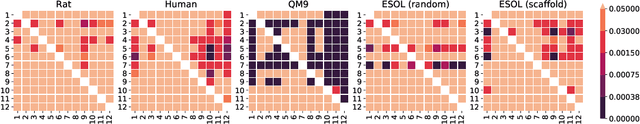
Graph neural networks have recently become a standard method for analysing chemical compounds. In the field of molecular property prediction, the emphasis is now put on designing new model architectures, and the importance of atom featurisation is oftentimes belittled. When contrasting two graph neural networks, the use of different atom features possibly leads to the incorrect attribution of the results to the network architecture. To provide a better understanding of this issue, we compare multiple atom representations for graph models and evaluate them on the prediction of free energy, solubility, and metabolic stability. To the best of our knowledge, this is the first methodological study that focuses on the relevance of atom representation to the predictive performance of graph neural networks.
Processing of incomplete images by (graph) convolutional neural networks
Oct 26, 2020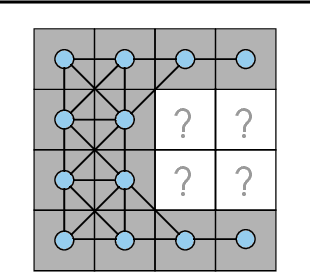
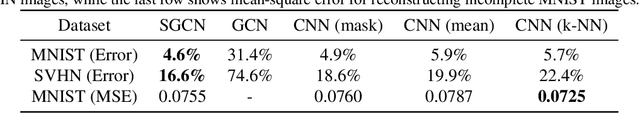
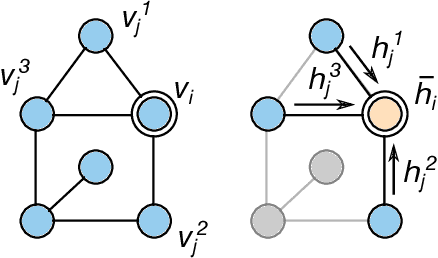

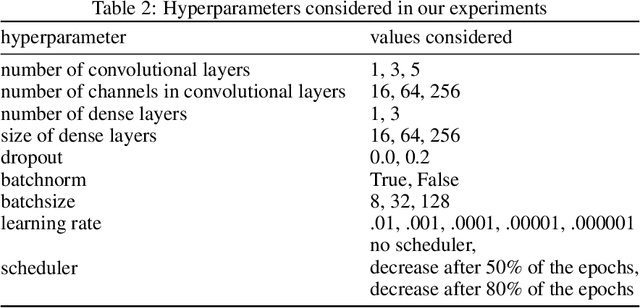
We investigate the problem of training neural networks from incomplete images without replacing missing values. For this purpose, we first represent an image as a graph, in which missing pixels are entirely ignored. The graph image representation is processed using a spatial graph convolutional network (SGCN) -- a type of graph convolutional networks, which is a proper generalization of classical CNNs operating on images. On one hand, our approach avoids the problem of missing data imputation while, on the other hand, there is a natural correspondence between CNNs and SGCN. Experiments confirm that our approach performs better than analogical CNNs with the imputation of missing values on typical classification and reconstruction tasks.
Flow-based anomaly detection
Oct 06, 2020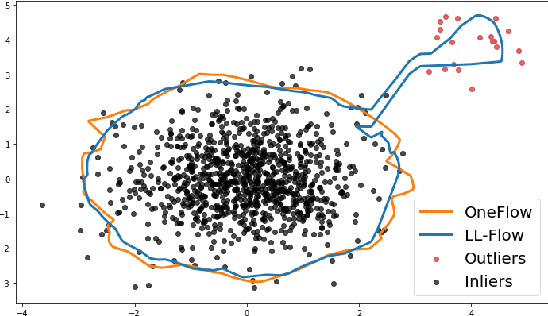
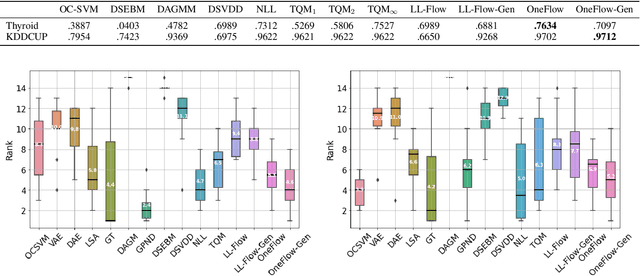
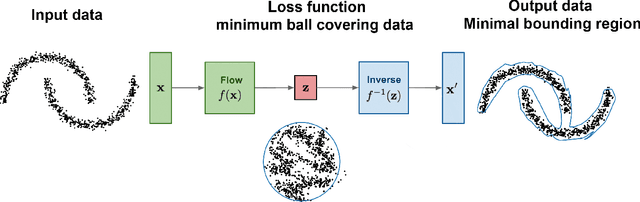
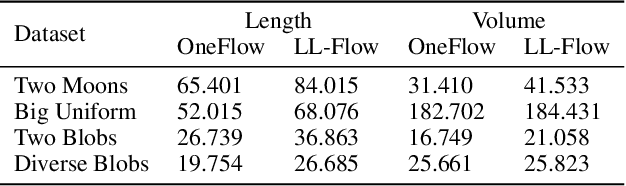
We propose OneFlow - a flow-based one-class classifier for anomaly (outliers) detection that finds a minimal volume bounding region. Contrary to density-based methods, OneFlow is constructed in such a way that its result typically does not depend on the structure of outliers. This is caused by the fact that during training the gradient of the cost function is propagated only over the points located near to the decision boundary (behavior similar to the support vectors in SVM). The combination of flow models and Bernstein quantile estimator allows OneFlow to find a parametric form of bounding region, which can be useful in various applications including describing shapes from 3D point clouds. Experiments show that the proposed model outperforms related methods on real-world anomaly detection problems.
Molecule Attention Transformer
Feb 19, 2020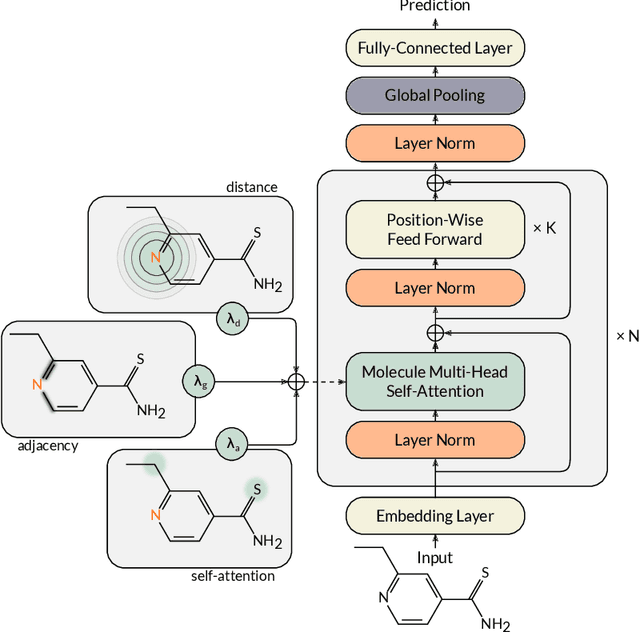

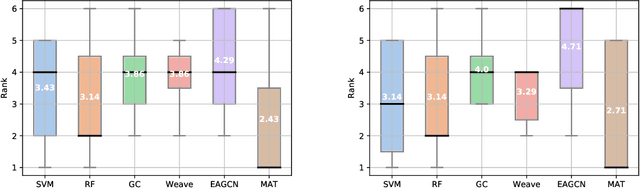
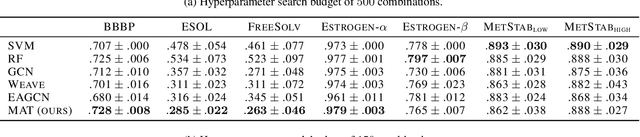
Designing a single neural network architecture that performs competitively across a range of molecule property prediction tasks remains largely an open challenge, and its solution may unlock a widespread use of deep learning in the drug discovery industry. To move towards this goal, we propose Molecule Attention Transformer (MAT). Our key innovation is to augment the attention mechanism in Transformer using inter-atomic distances and the molecular graph structure. Experiments show that MAT performs competitively on a diverse set of molecular prediction tasks. Most importantly, with a simple self-supervised pretraining, MAT requires tuning of only a few hyperparameter values to achieve state-of-the-art performance on downstream tasks. Finally, we show that attention weights learned by MAT are interpretable from the chemical point of view.
Geometric Graph Convolutional Neural Networks
Sep 11, 2019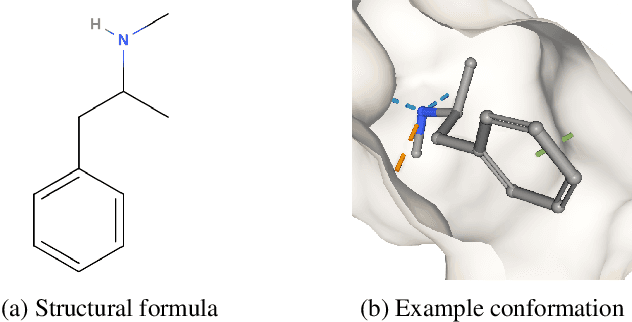
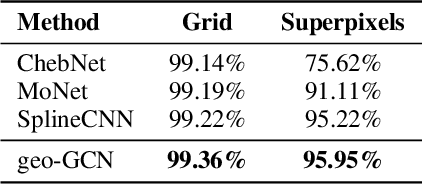
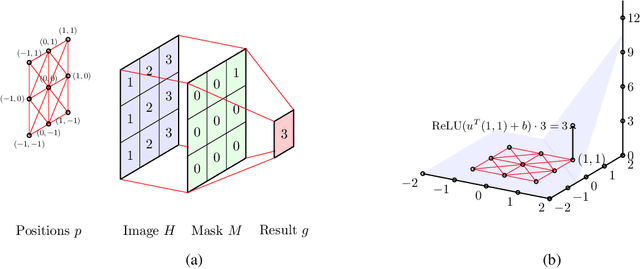
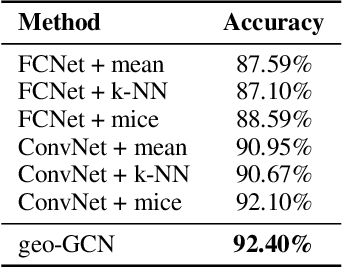
Graph Convolutional Networks (GCNs) have recently become the primary choice for learning from graph-structured data, superseding hash fingerprints in representing chemical compounds. However, GCNs lack the ability to take into account the ordering of node neighbors, even when there is a geometric interpretation of the graph vertices that provides an order based on their spatial positions. To remedy this issue, we propose Geometric Graph Convolutional Network (geo-GCN) which uses spatial features to efficiently learn from graphs that can be naturally located in space. Our contribution is threefold: we propose a GCN-inspired architecture which (i) leverages node positions, (ii) is a proper generalisation of both GCNs and Convolutional Neural Networks (CNNs), (iii) benefits from augmentation which further improves the performance and assures invariance with respect to the desired properties. Empirically, geo-GCN outperforms state-of-the-art graph-based methods on image classification and chemical tasks.