 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecule Attention Transformer
Feb 19, 2020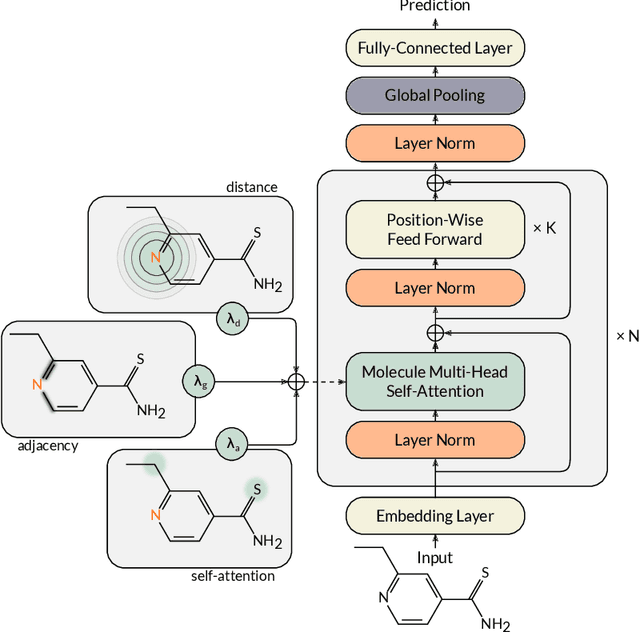

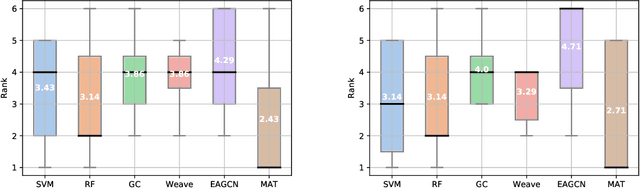
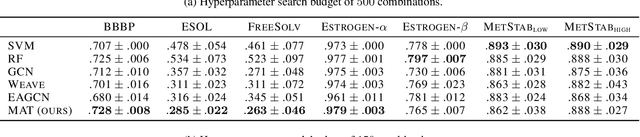
Designing a single neural network architecture that performs competitively across a range of molecule property prediction tasks remains largely an open challenge, and its solution may unlock a widespread use of deep learning in the drug discovery industry. To move towards this goal, we propose Molecule Attention Transformer (MAT). Our key innovation is to augment the attention mechanism in Transformer using inter-atomic distances and the molecular graph structure. Experiments show that MAT performs competitively on a diverse set of molecular prediction tasks. Most importantly, with a simple self-supervised pretraining, MAT requires tuning of only a few hyperparameter values to achieve state-of-the-art performance on downstream tasks. Finally, we show that attention weights learned by MAT are interpretable from the chemical point of view.
Mol-CycleGAN - a generative model for molecular optimization
Feb 06, 2019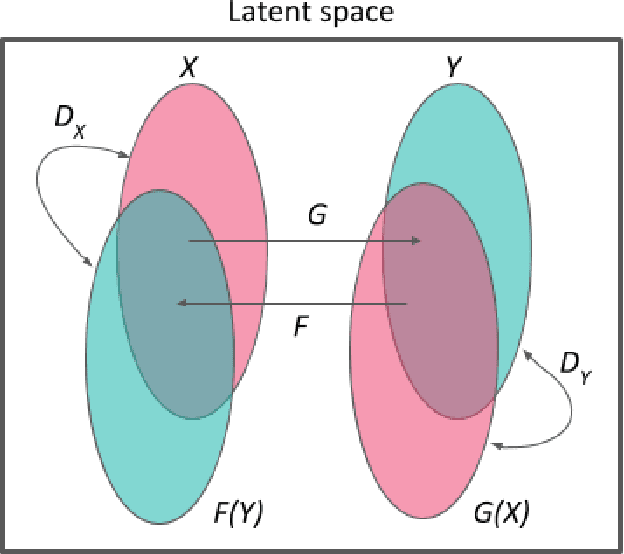

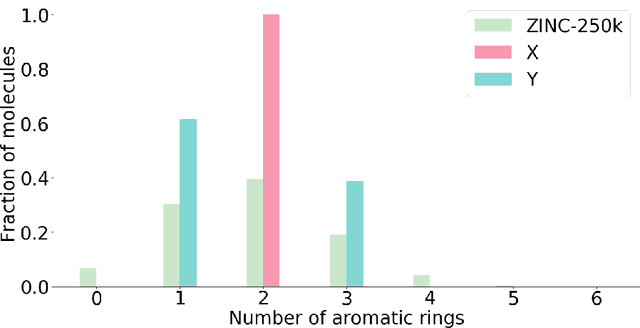
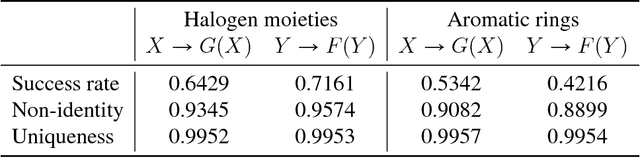
Designing a molecule with desired properties is one of the biggest challenges in drug development, as it requires optimization of chemical compound structures with respect to many complex properties. To augment the compound design process we introduce Mol-CycleGAN - a CycleGAN-based model that generates optimized compounds with high structural similarity to the original ones. Namely, given a molecule our model generates a structurally similar one with an optimized value of the considered property. We evaluate the performance of the model on selected optimization objectives related to structural properties (presence of halogen groups, number of aromatic rings) and to a physicochemical property (penalized logP). In the task of optimization of penalized logP of drug-like molecules our model significantly outperforms previous results.