 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLow cost prediction of probability distributions of molecular properties for early virtual screening
Jul 21, 2022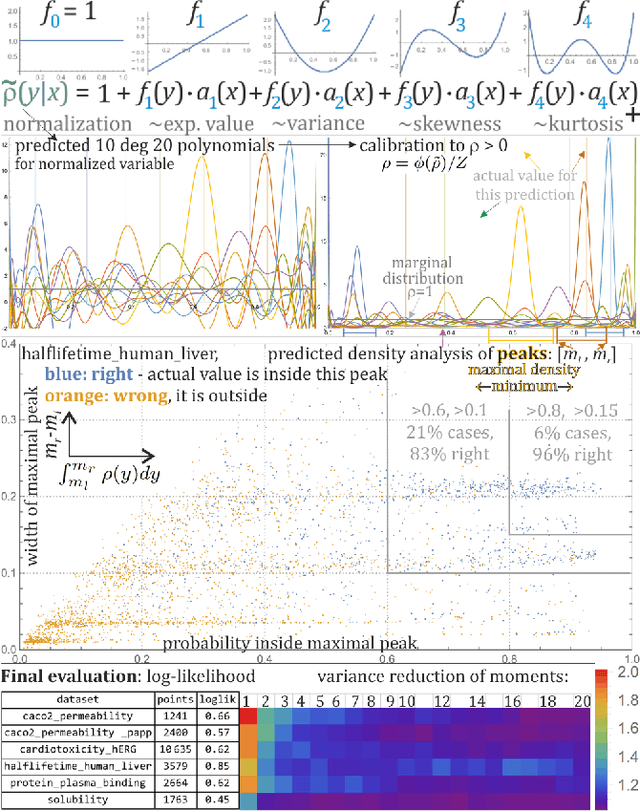
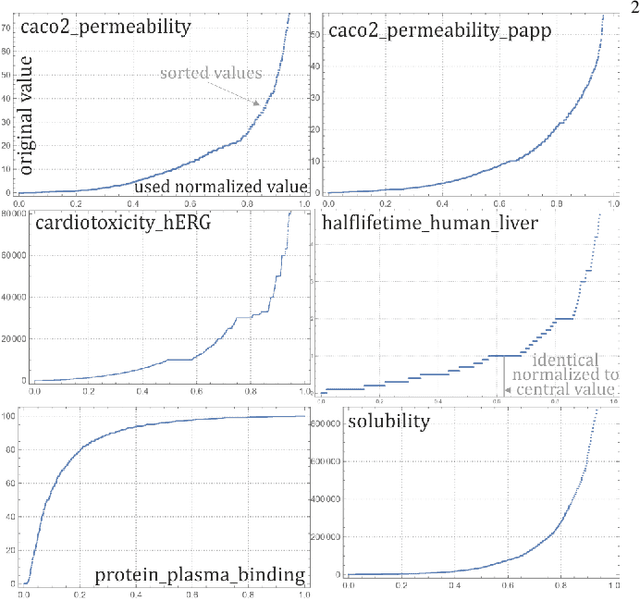


While there is a general focus on predictions of values, mathematically more appropriate is prediction of probability distributions: with additional possibilities like prediction of uncertainty, higher moments and quantiles. For the purpose of the computer-aided drug design field, this article applies Hierarchical Correlation Reconstruction approach, previously applied in the analysis of demographic, financial and astronomical data. Instead of a single linear regression to predict values, it uses multiple linear regressions to independently predict multiple moments, finally combining them into predicted probability distribution, here of several ADMET properties based on substructural fingerprint developed by Klekota\&Roth. Discussed application example is inexpensive selection of a percentage of molecules with properties nearly certain to be in a predicted or chosen range during virtual screening. Such an approach can facilitate the interpretation of the results as the predictions characterized by high rate of uncertainty are automatically detected. In addition, for each of the investigated predictive problems, we detected crucial structural features, which should be carefully considered when optimizing compounds towards particular property. The whole methodology developed in the study constitutes therefore a great support for medicinal chemists, as it enable fast rejection of compounds with the lowest potential of desired physicochemical/ADMET characteristic and guides the compound optimization process.
We Should at Least Be Able to Design Molecules That Dock Well
Jul 01, 2020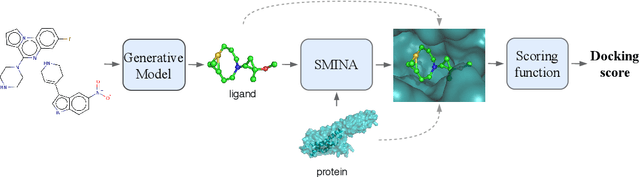
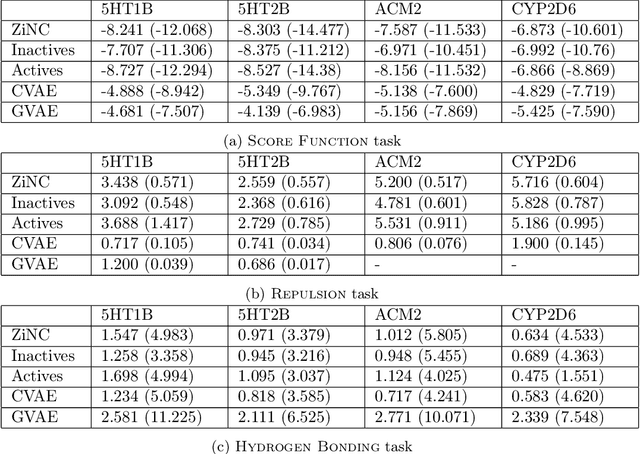
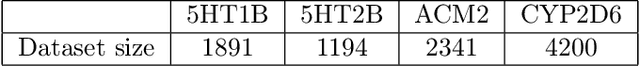
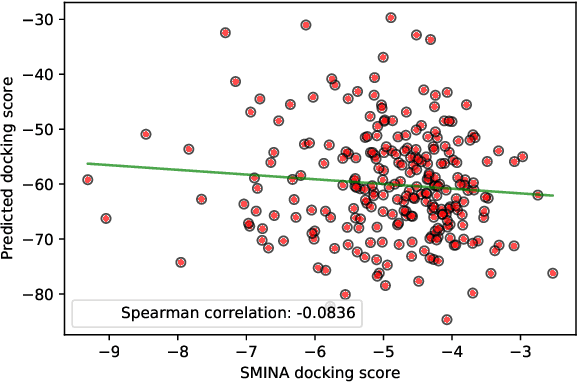
Designing compounds with desired properties is a key element of the drug discovery process. However, measuring progress in the field has been challenging due to the lack of realistic retrospective benchmarks, and the large cost of prospective validation. To close this gap, we propose a benchmark based on docking, a popular computational method for assessing molecule binding to a protein. Concretely, the goal is to generate drug-like molecules that are scored highly by SMINA, a popular docking software. We observe that popular graph-based generative models fail to generate molecules with a high docking score when trained using a realistically sized training set. This suggests a limitation of the current incarnation of models for de novo drug design. Finally, we propose a simplified version of the benchmark based on a simpler scoring function, and show that the tested models are able to partially solve it. We release the benchmark as an easy to use package available at https://github.com/cieplinski-tobiasz/smina-docking-benchmark. We hope that our benchmark will serve as a stepping stone towards the goal of automatically generating promising drug candidates.