 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDiAMoNDBack: Diffusion-denoising Autoregressive Model for Non-Deterministic Backmapping of Cα Protein Traces
Jul 23, 2023Coarse-grained molecular models of proteins permit access to length and time scales unattainable by all-atom models and the simulation of processes that occur on long-time scales such as aggregation and folding. The reduced resolution realizes computational accelerations but an atomistic representation can be vital for a complete understanding of mechanistic details. Backmapping is the process of restoring all-atom resolution to coarse-grained molecular models. In this work, we report DiAMoNDBack (Diffusion-denoising Autoregressive Model for Non-Deterministic Backmapping) as an autoregressive denoising diffusion probability model to restore all-atom details to coarse-grained protein representations retaining only C{\alpha} coordinates. The autoregressive generation process proceeds from the protein N-terminus to C-terminus in a residue-by-residue fashion conditioned on the C{\alpha} trace and previously backmapped backbone and side chain atoms within the local neighborhood. The local and autoregressive nature of our model makes it transferable between proteins. The stochastic nature of the denoising diffusion process means that the model generates a realistic ensemble of backbone and side chain all-atom configurations consistent with the coarse-grained C{\alpha} trace. We train DiAMoNDBack over 65k+ structures from Protein Data Bank (PDB) and validate it in applications to a hold-out PDB test set, intrinsically-disordered protein structures from the Protein Ensemble Database (PED), molecular dynamics simulations of fast-folding mini-proteins from DE Shaw Research, and coarse-grained simulation data. We achieve state-of-the-art reconstruction performance in terms of correct bond formation, avoidance of side chain clashes, and diversity of the generated side chain configurational states. We make DiAMoNDBack model publicly available as a free and open source Python package.
DEL-Dock: Molecular Docking-Enabled Modeling of DNA-Encoded Libraries
Dec 14, 2022DNA-Encoded Library (DEL) technology has enabled significant advances in hit identification by enabling efficient testing of combinatorially-generated molecular libraries. DEL screens measure protein binding affinity though sequencing reads of molecules tagged with unique DNA-barcodes that survive a series of selection experiments. Computational models have been deployed to learn the latent binding affinities that are correlated to the sequenced count data; however, this correlation is often obfuscated by various sources of noise introduced in its complicated data-generation process. In order to denoise DEL count data and screen for molecules with good binding affinity, computational models require the correct assumptions in their modeling structure to capture the correct signals underlying the data. Recent advances in DEL models have focused on probabilistic formulations of count data, but existing approaches have thus far been limited to only utilizing 2-D molecule-level representations. We introduce a new paradigm, DEL-Dock, that combines ligand-based descriptors with 3-D spatial information from docked protein-ligand complexes. 3-D spatial information allows our model to learn over the actual binding modality rather than using only structured-based information of the ligand. We show that our model is capable of effectively denoising DEL count data to predict molecule enrichment scores that are better correlated with experimental binding affinity measurements compared to prior works. Moreover, by learning over a collection of docked poses we demonstrate that our model, trained only on DEL data, implicitly learns to perform good docking pose selection without requiring external supervision from expensive-to-source protein crystal structures.
Discovery of Self-Assembling $π$-Conjugated Peptides by Active Learning-Directed Coarse-Grained Molecular Simulation
Jan 27, 2020
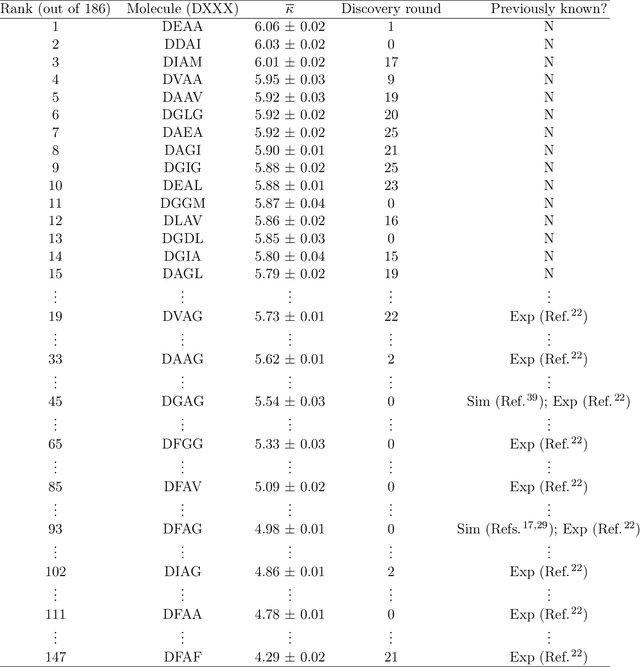


Electronically-active organic molecules have demonstrated great promise as novel soft materials for energy harvesting and transport. Self-assembled nanoaggregates formed from $\pi$-conjugated oligopeptides composed of an aromatic core flanked by oligopeptide wings offer emergent optoelectronic properties within a water soluble and biocompatible substrate. Nanoaggregate properties can be controlled by tuning core chemistry and peptide composition, but the sequence-structure-function relations remain poorly characterized. In this work, we employ coarse-grained molecular dynamics simulations within an active learning protocol employing deep representational learning and Bayesian optimization to efficiently identify molecules capable of assembling pseudo-1D nanoaggregates with good stacking of the electronically-active $\pi$-cores. We consider the DXXX-OPV3-XXXD oligopeptide family, where D is an Asp residue and OPV3 is an oligophenylene vinylene oligomer (1,4-distyrylbenzene), to identify the top performing XXX tripeptides within all 20$^3$ = 8,000 possible sequences. By direct simulation of only 2.3% of this space, we identify molecules predicted to exhibit superior assembly relative to those reported in prior work. Spectral clustering of the top candidates reveals new design rules governing assembly. This work establishes new understanding of DXXX-OPV3-XXXD assembly, identifies promising new candidates for experimental testing, and presents a computational design platform that can be generically extended to other peptide-based and peptide-like systems.