 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAssessing Sample Quality in Conditional Generation under Compositional Shift
Jun 09, 2026Conditional generators provide a natural tool for controllable generation, including settings where the desired condition is a new composition of observed attributes or experimental factors. In many applications, especially in scientific domains, such models are attractive to explore conditions for which real samples are rare, expensive, or not yet observed. However, this creates a circularity for evaluation: standard conditional quality metrics require a reference target distribution, but in the extrapolative regime that distribution is unavailable by definition. We address this problem with a post-hoc, per-sample trust score for assessing conditional samples using only the training distribution. The score combines two estimable quantities: global realism, measuring compatibility with the real data manifold, and attribute-wise faithfulness, measuring whether a sample is closer to the requested attributes than to plausible alternatives. We show that the score can recover meaningful comparisons across extrapolated generations, under a mild coverage condition on the observed attributes. These comparisons enable effective filtering, ranking, and abstention of generations and can be used directly on off-the-shelf pretrained models. In biological imaging, selected samples preserve real morphological structure better and improve downstream predictive performance, while similar gains are observed on controlled vision benchmarks. Finally, we show how the score can be applied during generation, enabling abstention before full decoding. Code is available at https://github.com/berkerdemirel/faithful-cond-gen.
Toward Identifiable Sparse Autoencoders
May 29, 2026Recently, sparse autoencoders (SAEs) have emerged as an attractive tool for interpreting and interacting with representations in practical neural networks. While it is common empirical folklore, we also show theoretically that SAEs are highly unstable: different training runs are likely to produce different concept dictionaries and sparse codes. We characterize the model properties that hinder the stability of real-world SAEs, and address each of these problems through minimal changes to the architecture and training procedure. Together, these changes yield two versions of an \textbf{i}dentifiable SAE (iSAE), a variant of the standard TopK SAE with lower reconstruction error and improved stability. We explain this improvement theoretically by connecting SAEs with traditional dictionary learning approaches, and show that the dictionaries learned in practice satisfy an approximate restricted isometry condition, rendering the corresponding sparse codes in those models near-identifiable.
Statistical and structural identifiability in representation learning
Mar 12, 2026Representation learning models exhibit a surprising stability in their internal representations. Whereas most prior work treats this stability as a single property, we formalize it as two distinct concepts: statistical identifiability (consistency of representations across runs) and structural identifiability (alignment of representations with some unobserved ground truth). Recognizing that perfect pointwise identifiability is generally unrealistic for modern representation learning models, we propose new model-agnostic definitions of statistical and structural near-identifiability of representations up to some error tolerance $ε$. Leveraging these definitions, we prove a statistical $ε$-near-identifiability result for the representations of models with nonlinear decoders, generalizing existing identifiability theory beyond last-layer representations in e.g. generative pre-trained transformers (GPTs) to near-identifiability of the intermediate representations of a broad class of models including (masked) autoencoders (MAEs) and supervised learners. Although these weaker assumptions confer weaker identifiability, we show that independent components analysis (ICA) can resolve much of the remaining linear ambiguity for this class of models, and validate and measure our near-identifiability claims empirically. With additional assumptions on the data-generating process, statistical identifiability extends to structural identifiability, yielding a simple and practical recipe for disentanglement: ICA post-processing of latent representations. On synthetic benchmarks, this approach achieves state-of-the-art disentanglement using a vanilla autoencoder. With a foundation model-scale MAE for cell microscopy, it disentangles biological variation from technical batch effects, substantially improving downstream generalization.
Calibrated Test-Time Guidance for Bayesian Inference
Feb 25, 2026Test-time guidance is a widely used mechanism for steering pretrained diffusion models toward outcomes specified by a reward function. Existing approaches, however, focus on maximizing reward rather than sampling from the true Bayesian posterior, leading to miscalibrated inference. In this work, we show that common test-time guidance methods do not recover the correct posterior distribution and identify the structural approximations responsible for this failure. We then propose consistent alternative estimators that enable calibrated sampling from the Bayesian posterior. We significantly outperform previous methods on a set of Bayesian inference tasks, and match state-of-the-art in black hole image reconstruction.
Parallel Token Prediction for Language Models
Dec 24, 2025We propose Parallel Token Prediction (PTP), a universal framework for parallel sequence generation in language models. PTP jointly predicts multiple dependent tokens in a single transformer call by incorporating the sampling procedure into the model. This reduces the latency bottleneck of autoregressive decoding, and avoids the restrictive independence assumptions common in existing multi-token prediction methods. We prove that PTP can represent arbitrary autoregressive sequence distributions. PTP is trained either by distilling an existing model or through inverse autoregressive training without a teacher. Experimentally, we achieve state-of-the-art speculative decoding performance on Vicuna-7B by accepting over four tokens per step on Spec-Bench. The universality of our framework indicates that parallel generation of long sequences is feasible without loss of modeling power.
How Well Do LLMs Understand Drug Mechanisms? A Knowledge + Reasoning Evaluation Dataset
Nov 09, 2025Two scientific fields showing increasing interest in pre-trained large language models (LLMs) are drug development / repurposing, and personalized medicine. For both, LLMs have to demonstrate factual knowledge as well as a deep understanding of drug mechanisms, so they can recall and reason about relevant knowledge in novel situations. Drug mechanisms of action are described as a series of interactions between biomedical entities, which interlink into one or more chains directed from the drug to the targeted disease. Composing the effects of the interactions in a candidate chain leads to an inference about whether the drug might be useful or not for that disease. We introduce a dataset that evaluates LLMs on both factual knowledge of known mechanisms, and their ability to reason about them under novel situations, presented as counterfactuals that the models are unlikely to have seen during training. Using this dataset, we show that o4-mini outperforms the 4o, o3, and o3-mini models from OpenAI, and the recent small Qwen3-4B-thinking model closely matches o4-mini's performance, even outperforming it in some cases. We demonstrate that the open world setting for reasoning tasks, which requires the model to recall relevant knowledge, is more challenging than the closed world setting where the needed factual knowledge is provided. We also show that counterfactuals affecting internal links in the reasoning chain present a much harder task than those affecting a link from the drug mentioned in the prompt.
Variational Control for Guidance in Diffusion Models
Feb 06, 2025



Diffusion models exhibit excellent sample quality, but existing guidance methods often require additional model training or are limited to specific tasks. We revisit guidance in diffusion models from the perspective of variational inference and control, introducing Diffusion Trajectory Matching (DTM) that enables guiding pretrained diffusion trajectories to satisfy a terminal cost. DTM unifies a broad class of guidance methods and enables novel instantiations. We introduce a new method within this framework that achieves state-of-the-art results on several linear and (blind) non-linear inverse problems without requiring additional model training or modifications. For instance, in ImageNet non-linear deblurring, our model achieves an FID score of 34.31, significantly improving over the best pretrained-method baseline (FID 78.07). We will make the code available in a future update.
Adjusting Pretrained Backbones for Performativity
Oct 06, 2024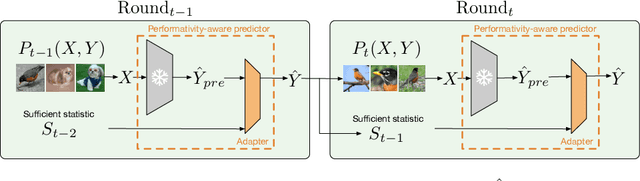



With the widespread deployment of deep learning models, they influence their environment in various ways. The induced distribution shifts can lead to unexpected performance degradation in deployed models. Existing methods to anticipate performativity typically incorporate information about the deployed model into the feature vector when predicting future outcomes. While enjoying appealing theoretical properties, modifying the input dimension of the prediction task is often not practical. To address this, we propose a novel technique to adjust pretrained backbones for performativity in a modular way, achieving better sample efficiency and enabling the reuse of existing deep learning assets. Focusing on performative label shift, the key idea is to train a shallow adapter module to perform a Bayes-optimal label shift correction to the backbone's logits given a sufficient statistic of the model to be deployed. As such, our framework decouples the construction of input-specific feature embeddings from the mechanism governing performativity. Motivated by dynamic benchmarking as a use-case, we evaluate our approach under adversarial sampling, for vision and language tasks. We show how it leads to smaller loss along the retraining trajectory and enables us to effectively select among candidate models to anticipate performance degradations. More broadly, our work provides a first baseline for addressing performativity in deep learning.
How to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities
Sep 18, 2024

The cell is arguably the smallest unit of life and is central to understanding biology. Accurate modeling of cells is important for this understanding as well as for determining the root causes of disease. Recent advances in artificial intelligence (AI), combined with the ability to generate large-scale experimental data, present novel opportunities to model cells. Here we propose a vision of AI-powered Virtual Cells, where robust representations of cells and cellular systems under different conditions are directly learned from growing biological data across measurements and scales. We discuss desired capabilities of AI Virtual Cells, including generating universal representations of biological entities across scales, and facilitating interpretable in silico experiments to predict and understand their behavior using Virtual Instruments. We further address the challenges, opportunities and requirements to realize this vision including data needs, evaluation strategies, and community standards and engagement to ensure biological accuracy and broad utility. We envision a future where AI Virtual Cells help identify new drug targets, predict cellular responses to perturbations, as well as scale hypothesis exploration. With open science collaborations across the biomedical ecosystem that includes academia, philanthropy, and the biopharma and AI industries, a comprehensive predictive understanding of cell mechanisms and interactions is within reach.
Position Paper: Bayesian Deep Learning in the Age of Large-Scale AI
Feb 06, 2024
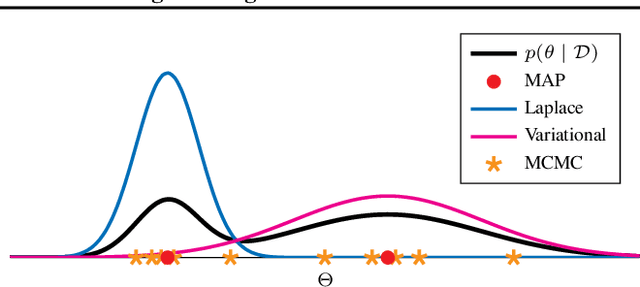
In the current landscape of deep learning research, there is a predominant emphasis on achieving high predictive accuracy in supervised tasks involving large image and language datasets. However, a broader perspective reveals a multitude of overlooked metrics, tasks, and data types, such as uncertainty, active and continual learning, and scientific data, that demand attention. Bayesian deep learning (BDL) constitutes a promising avenue, offering advantages across these diverse settings. This paper posits that BDL can elevate the capabilities of deep learning. It revisits the strengths of BDL, acknowledges existing challenges, and highlights some exciting research avenues aimed at addressing these obstacles. Looking ahead, the discussion focuses on possible ways to combine large-scale foundation models with BDL to unlock their full potential.