Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePath-Augmented Graph Transformer Network
Paper and Code
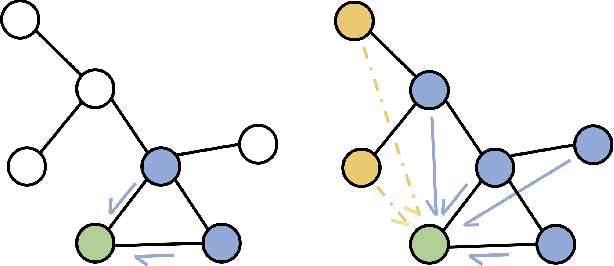
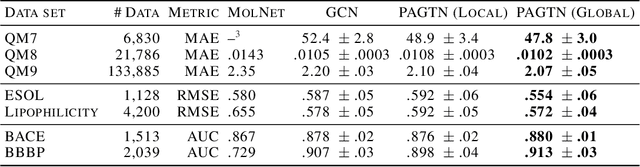


Much of the recent work on learning molecular representations has been based on Graph Convolution Networks (GCN). These models rely on local aggregation operations and can therefore miss higher-order graph properties. To remedy this, we propose Path-Augmented Graph Transformer Networks (PAGTN) that are explicitly built on longer-range dependencies in graph-structured data. Specifically, we use path features in molecular graphs to create global attention layers. We compare our PAGTN model against the GCN model and show that our model consistently outperforms GCNs on molecular property prediction datasets including quantum chemistry (QM7, QM8, QM9), physical chemistry (ESOL, Lipophilictiy) and biochemistry (BACE, BBBP).
* Appears in ICML LRG Workshop
View paper on