 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnderstanding Protein Dynamics with L1-Regularized Reversible Hidden Markov Models
Paper and Code
May 06, 2014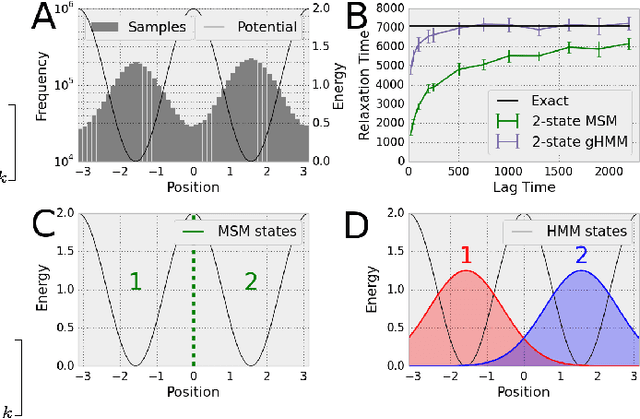
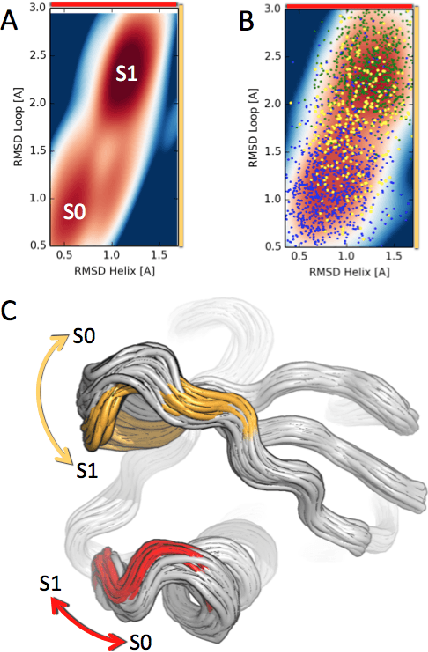
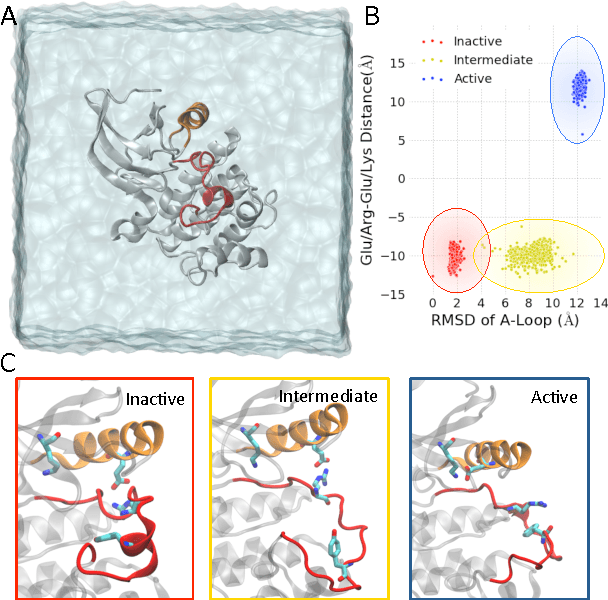
We present a machine learning framework for modeling protein dynamics. Our approach uses L1-regularized, reversible hidden Markov models to understand large protein datasets generated via molecular dynamics simulations. Our model is motivated by three design principles: (1) the requirement of massive scalability; (2) the need to adhere to relevant physical law; and (3) the necessity of providing accessible interpretations, critical for both cellular biology and rational drug design. We present an EM algorithm for learning and introduce a model selection criteria based on the physical notion of convergence in relaxation timescales. We contrast our model with standard methods in biophysics and demonstrate improved robustness. We implement our algorithm on GPUs and apply the method to two large protein simulation datasets generated respectively on the NCSA Bluewaters supercomputer and the Folding@Home distributed computing network. Our analysis identifies the conformational dynamics of the ubiquitin protein critical to cellular signaling, and elucidates the stepwise activation mechanism of the c-Src kinase protein.