 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredictive models of RNA degradation through dual crowdsourcing
Oct 14, 2021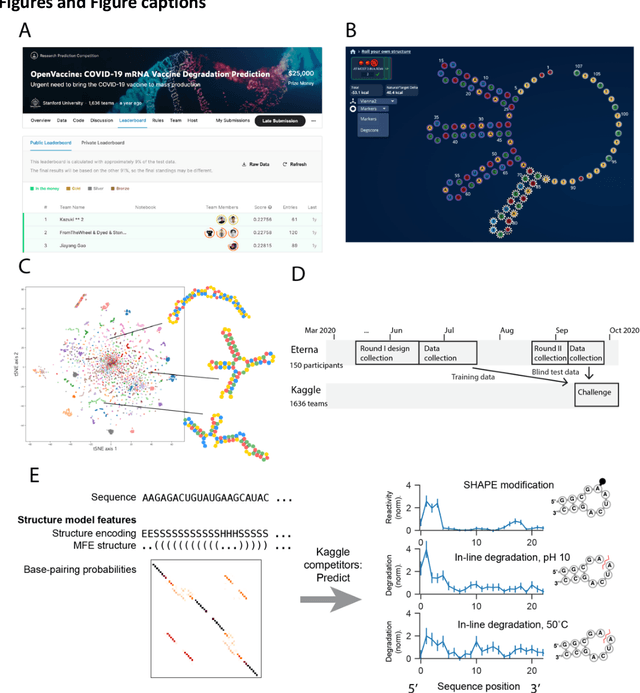
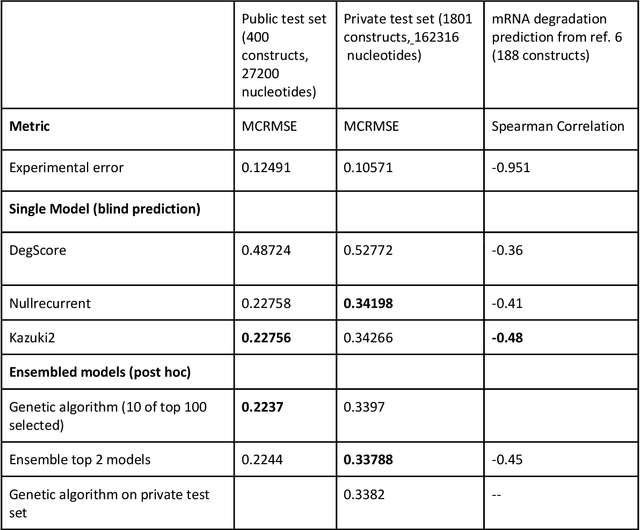
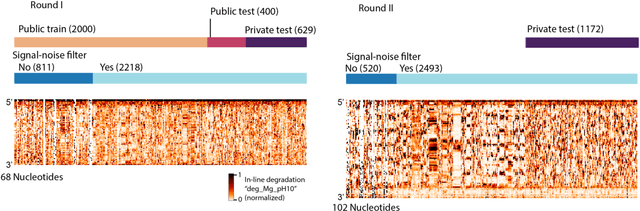
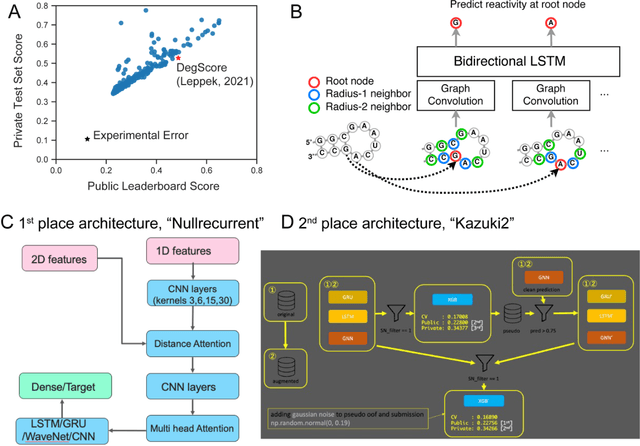
Messenger RNA-based medicines hold immense potential, as evidenced by their rapid deployment as COVID-19 vaccines. However, worldwide distribution of mRNA molecules has been limited by their thermostability, which is fundamentally limited by the intrinsic instability of RNA molecules to a chemical degradation reaction called in-line hydrolysis. Predicting the degradation of an RNA molecule is a key task in designing more stable RNA-based therapeutics. Here, we describe a crowdsourced machine learning competition ("Stanford OpenVaccine") on Kaggle, involving single-nucleotide resolution measurements on 6043 102-130-nucleotide diverse RNA constructs that were themselves solicited through crowdsourcing on the RNA design platform Eterna. The entire experiment was completed in less than 6 months. Winning models demonstrated test set errors that were better by 50% than the previous state-of-the-art DegScore model. Furthermore, these models generalized to blindly predicting orthogonal degradation data on much longer mRNA molecules (504-1588 nucleotides) with improved accuracy over DegScore and other models. Top teams integrated natural language processing architectures and data augmentation techniques with predictions from previous dynamic programming models for RNA secondary structure. These results indicate that such models are capable of representing in-line hydrolysis with excellent accuracy, supporting their use for designing stabilized messenger RNAs. The integration of two crowdsourcing platforms, one for data set creation and another for machine learning, may be fruitful for other urgent problems that demand scientific discovery on rapid timescales.
Note: Variational Encoding of Protein Dynamics Benefits from Maximizing Latent Autocorrelation
Mar 17, 2018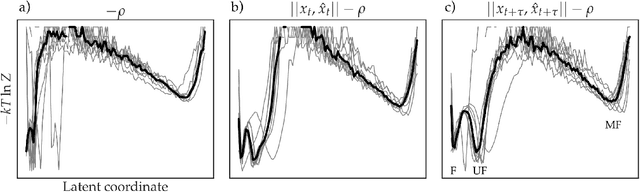
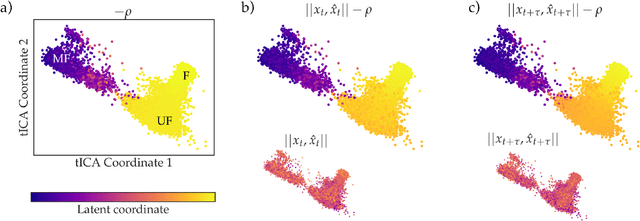
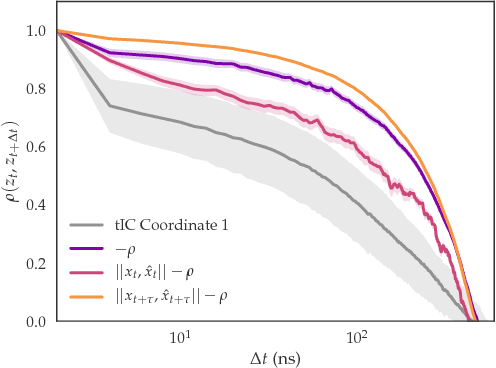
As deep Variational Auto-Encoder (VAE) frameworks become more widely used for modeling biomolecular simulation data, we emphasize the capability of the VAE architecture to concurrently maximize the timescale of the latent space while inferring a reduced coordinate, which assists in finding slow processes as according to the variational approach to conformational dynamics. We additionally provide evidence that the VDE framework (Hern\'andez et al., 2017), which uses this autocorrelation loss along with a time-lagged reconstruction loss, obtains a variationally optimized latent coordinate in comparison with related loss functions. We thus recommend leveraging the autocorrelation of the latent space while training neural network models of biomolecular simulation data to better represent slow processes.
Transferable neural networks for enhanced sampling of protein dynamics
Jan 02, 2018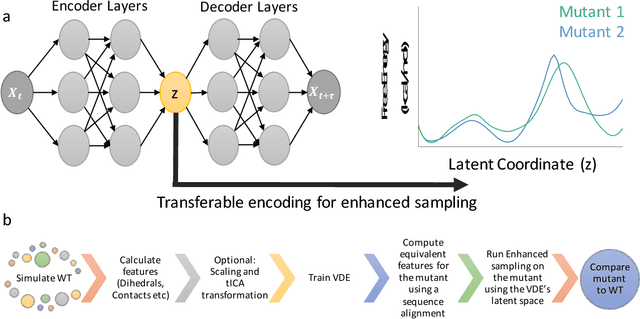
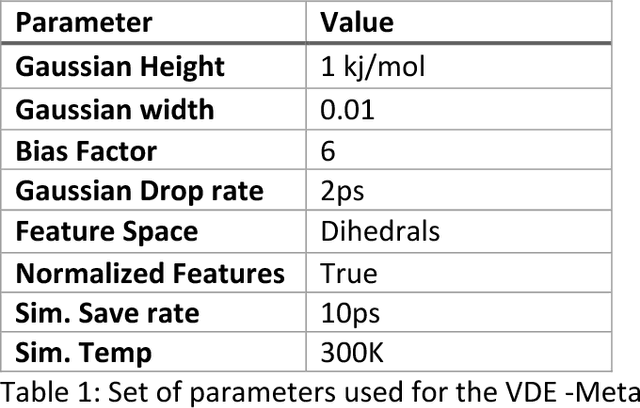
Variational auto-encoder frameworks have demonstrated success in reducing complex nonlinear dynamics in molecular simulation to a single non-linear embedding. In this work, we illustrate how this non-linear latent embedding can be used as a collective variable for enhanced sampling, and present a simple modification that allows us to rapidly perform sampling in multiple related systems. We first demonstrate our method is able to describe the effects of force field changes in capped alanine dipeptide after learning a model using AMBER99. We further provide a simple extension to variational dynamics encoders that allows the model to be trained in a more efficient manner on larger systems by encoding the outputs of a linear transformation using time-structure based independent component analysis (tICA). Using this technique, we show how such a model trained for one protein, the WW domain, can efficiently be transferred to perform enhanced sampling on a related mutant protein, the GTT mutation. This method shows promise for its ability to rapidly sample related systems using a single transferable collective variable and is generally applicable to sets of related simulations, enabling us to probe the effects of variation in increasingly large systems of biophysical interest.
Variational Encoding of Complex Dynamics
Dec 02, 2017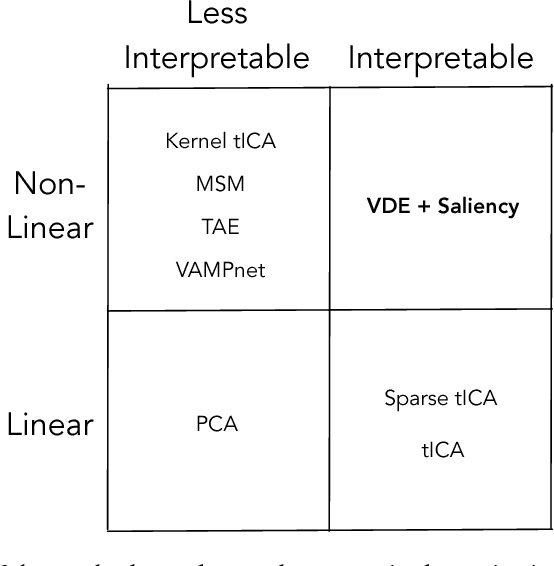
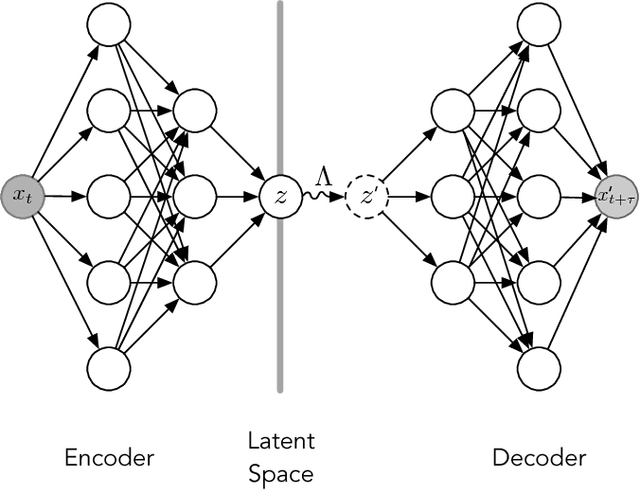
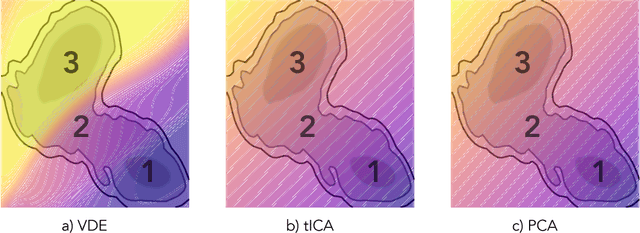
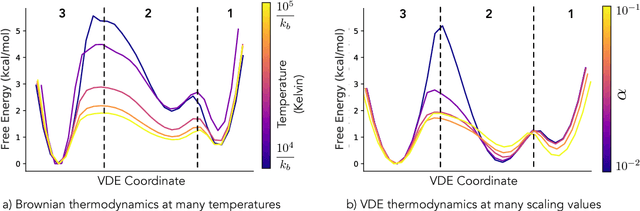
Often the analysis of time-dependent chemical and biophysical systems produces high-dimensional time-series data for which it can be difficult to interpret which individual features are most salient. While recent work from our group and others has demonstrated the utility of time-lagged co-variate models to study such systems, linearity assumptions can limit the compression of inherently nonlinear dynamics into just a few characteristic components. Recent work in the field of deep learning has led to the development of variational autoencoders (VAE), which are able to compress complex datasets into simpler manifolds. We present the use of a time-lagged VAE, or variational dynamics encoder (VDE), to reduce complex, nonlinear processes to a single embedding with high fidelity to the underlying dynamics. We demonstrate how the VDE is able to capture nontrivial dynamics in a variety of examples, including Brownian dynamics and atomistic protein folding. Additionally, we demonstrate a method for analyzing the VDE model, inspired by saliency mapping, to determine what features are selected by the VDE model to describe dynamics. The VDE presents an important step in applying techniques from deep learning to more accurately model and interpret complex biophysics.
* Fixed typos and added references