 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomated design of collective variables using supervised machine learning
Paper and Code
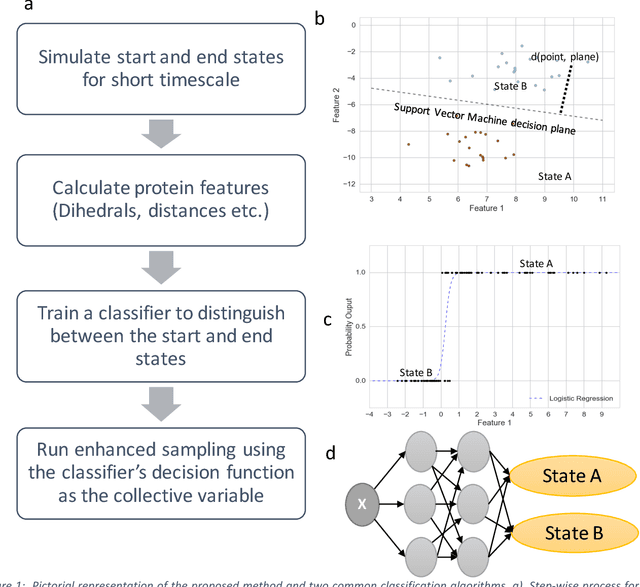
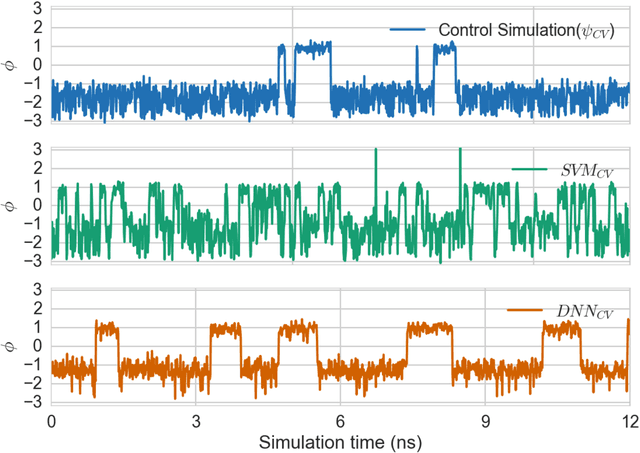
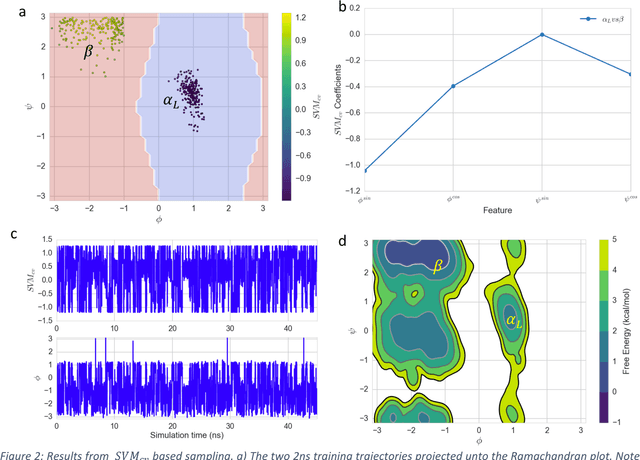

Selection of appropriate collective variables for enhancing sampling of molecular simulations remains an unsolved problem in computational biophysics. In particular, picking initial collective variables (CVs) is particularly challenging in higher dimensions. Which atomic coordinates or transforms there of from a list of thousands should one pick for enhanced sampling runs? How does a modeler even begin to pick starting coordinates for investigation? This remains true even in the case of simple two state systems and only increases in difficulty for multi-state systems. In this work, we solve the initial CV problem using a data-driven approach inspired by the filed of supervised machine learning. In particular, we show how the decision functions in supervised machine learning (SML) algorithms can be used as initial CVs (SML_cv) for accelerated sampling. Using solvated alanine dipeptide and Chignolin mini-protein as our test cases, we illustrate how the distance to the Support Vector Machines' decision hyperplane, the output probability estimates from Logistic Regression, the outputs from deep neural network classifiers, and other classifiers may be used to reversibly sample slow structural transitions. We discuss the utility of other SML algorithms that might be useful for identifying CVs for accelerating molecular simulations.