 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePre-training Graph Neural Networks
Paper and Code
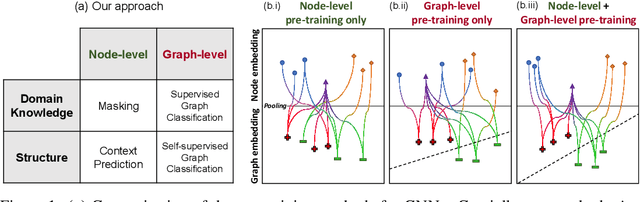
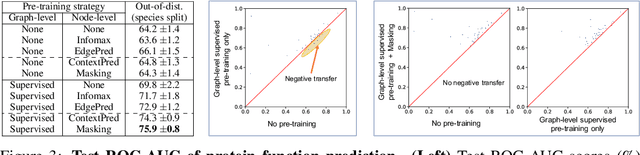
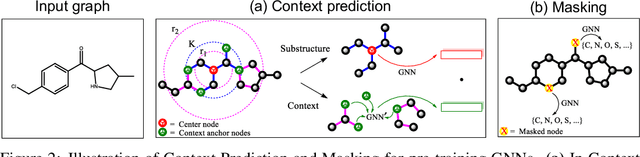
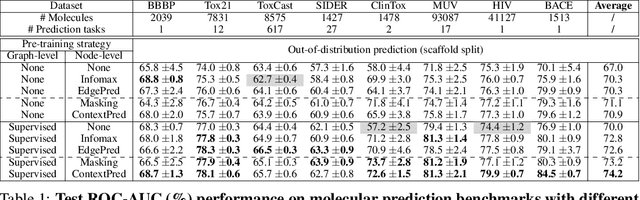
Many applications of machine learning in science and medicine, including molecular property and protein function prediction, can be cast as problems of predicting some properties of graphs, where having good graph representations is critical. However, two key challenges in these domains are (1) extreme scarcity of labeled data due to expensive lab experiments, and (2) needing to extrapolate to test graphs that are structurally different from those seen during training. In this paper, we explore pre-training to address both of these challenges. In particular, working with Graph Neural Networks (GNNs) for representation learning of graphs, we wish to obtain node representations that (1) capture similarity of nodes' network neighborhood structure, (2) can be composed to give accurate graph-level representations, and (3) capture domain-knowledge. To achieve these goals, we propose a series of methods to pre-train GNNs at both the node-level and the graph-level, using both unlabeled data and labeled data from related auxiliary supervised tasks. We perform extensive evaluation on two applications, molecular property and protein function prediction. We observe that performing only graph-level supervised pre-training often leads to marginal performance gain or even can worsen the performance compared to non-pre-trained models. On the other hand, effectively combining both node- and graph-level pre-training techniques significantly improves generalization to out-of-distribution graphs, consistently outperforming non-pre-trained GNNs across 8 datasets in molecular property prediction (resp. 40 tasks in protein function prediction), with the average ROC-AUC improvement of 7.2% (resp. 11.7%).