 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCrystal Structure Search with Random Relaxations Using Graph Networks
Dec 08, 2020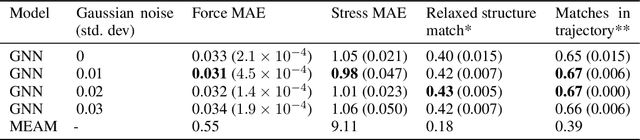
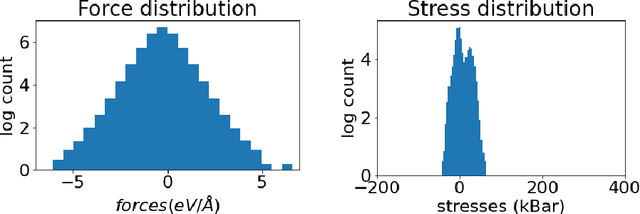
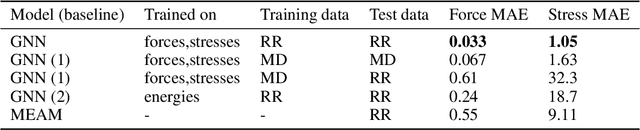
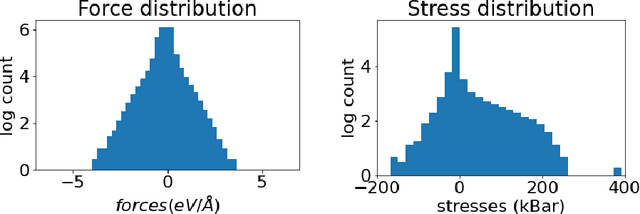
Materials design enables technologies critical to humanity, including combating climate change with solar cells and batteries. Many properties of a material are determined by its atomic crystal structure. However, prediction of the atomic crystal structure for a given material's chemical formula is a long-standing grand challenge that remains a barrier in materials design. We investigate a data-driven approach to accelerating ab initio random structure search (AIRSS), a state-of-the-art method for crystal structure search. We build a novel dataset of random structure relaxations of Li-Si battery anode materials using high-throughput density functional theory calculations. We train graph neural networks to simulate relaxations of random structures. Our model is able to find an experimentally verified structure of Li15Si4 it was not trained on, and has potential for orders of magnitude speedup over AIRSS when searching large unit cells and searching over multiple chemical stoichiometries. Surprisingly, we find that data augmentation of adding Gaussian noise improves both the accuracy and out of domain generalization of our models.