 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhase discovery with active learning: Application to structural phase transitions in equiatomic NiTi
Jan 10, 2024Nickel titanium (NiTi) is a protypical shape-memory alloy used in a range of biomedical and engineering devices, but direct molecular dynamics simulations of the martensitic B19' -> B2 phase transition driving its shape-memory behavior are rare and have relied on classical force fields with limited accuracy. Here, we train four machine-learned force fields for equiatomic NiTi based on the LDA, PBE, PBEsol, and SCAN DFT functionals. The models are trained on the fly during NPT molecular dynamics, with DFT calculations and model updates performed automatically whenever the uncertainty of a local energy prediction exceeds a chosen threshold. The models achieve accuracies of 1-2 meV/atom during training and are shown to closely track DFT predictions of B2 and B19' elastic constants and phonon frequencies. Surprisingly, in large-scale molecular dynamics simulations, only the SCAN model predicts a reversible B19' -> B2 phase transition, with the LDA, PBE, and PBEsol models predicting a reversible transition to a previously uncharacterized low-volume phase, which we hypothesize to be a new stable high-pressure phase. We examine the structure of the new phase and estimate its stability on the temperature-pressure phase diagram. This work establishes an automated active learning protocol for studying displacive transformations, reveals important differences between DFT functionals that can only be detected in large-scale simulations, provides an accurate force field for NiTi, and identifies a new phase.
Multitask machine learning of collective variables for enhanced sampling of rare events
Dec 07, 2020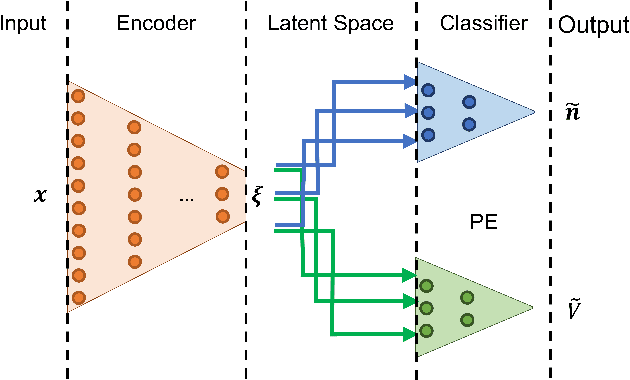
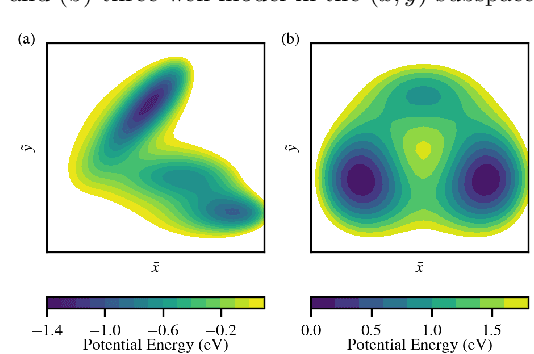
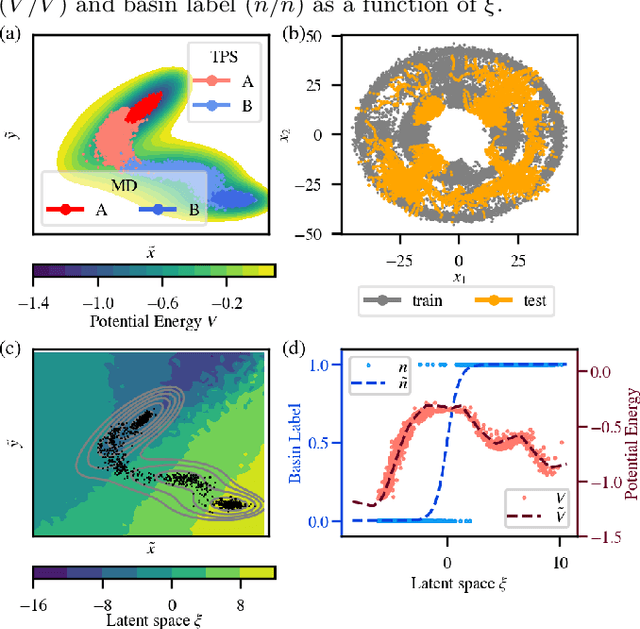
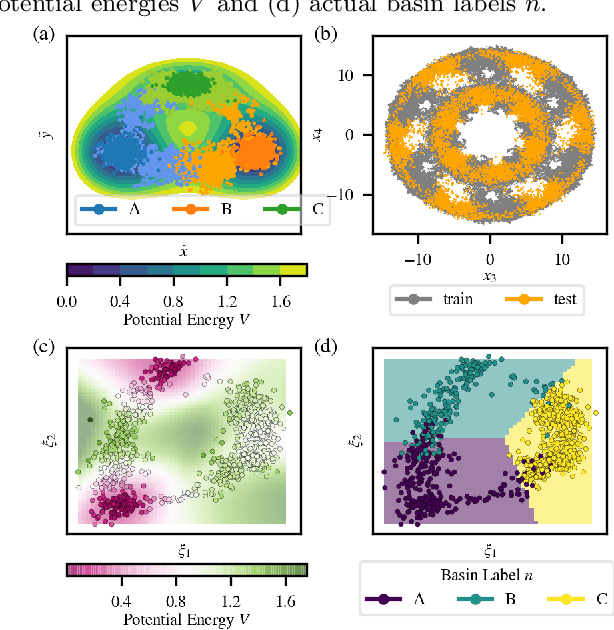
Computing accurate reaction rates is a central challenge in computational chemistry and biology because of the high cost of free energy estimation with unbiased molecular dynamics. In this work, a data-driven machine learning algorithm is devised to learn collective variables with a multitask neural network, where a common upstream part reduces the high dimensionality of atomic configurations to a low dimensional latent space, and separate downstream parts map the latent space to predictions of basin class labels and potential energies. The resulting latent space is shown to be an effective low-dimensional representation, capturing the reaction progress and guiding effective umbrella sampling to obtain accurate free energy landscapes. This approach is successfully applied to model systems including a 5D M\"uller Brown model, a 5D three-well model, and alanine dipeptide in vacuum. This approach enables automated dimensionality reduction for energy controlled reactions in complex systems, offers a unified framework that can be trained with limited data, and outperforms single-task learning approaches, including autoencoders.
Fast Bayesian Force Fields from Active Learning: Study of Inter-Dimensional Transformation of Stanene
Aug 26, 2020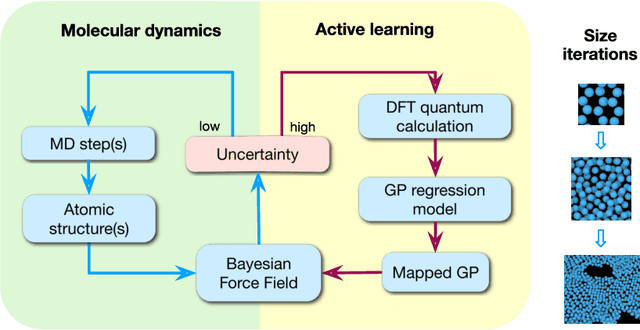
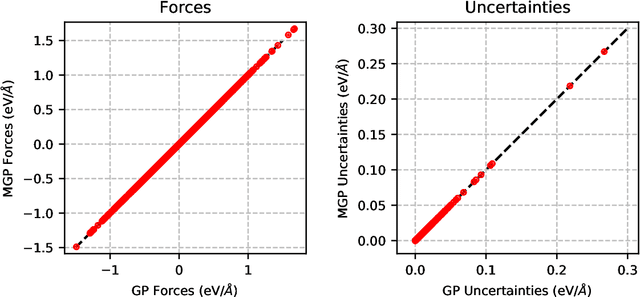
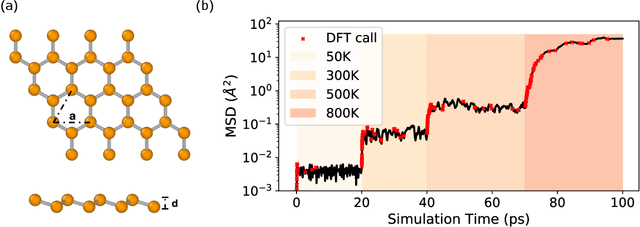
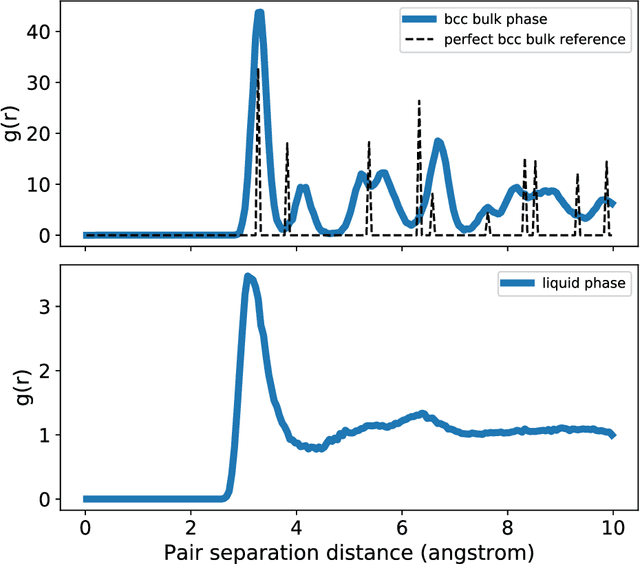
We present a way to dramatically accelerate Gaussian process models for interatomic force fields based on many-body kernels by mapping both forces and uncertainties onto functions of low-dimensional features. This allows for automated active learning of models combining near-quantum accuracy, built-in uncertainty, and constant cost of evaluation that is comparable to classical analytical models, capable of simulating millions of atoms. Using this approach, we perform large scale molecular dynamics simulations of the stability of the stanene monolayer. We discover an unusual phase transformation mechanism of 2D stanene, where ripples lead to nucleation of bilayer defects, densification into a disordered multilayer structure, followed by formation of bulk liquid at high temperature or nucleation and growth of the 3D bcc crystal at low temperature. The presented method opens possibilities for rapid development of fast accurate uncertainty-aware models for simulating long-time large-scale dynamics of complex materials.