 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHigh-performance training and inference for deep equivariant interatomic potentials
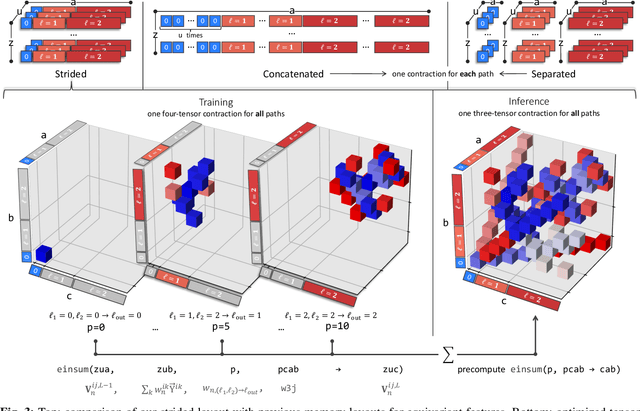
Apr 22, 2025Machine learning interatomic potentials, particularly those based on deep equivariant neural networks, have demonstrated state-of-the-art accuracy and computational efficiency in atomistic modeling tasks like molecular dynamics and high-throughput screening. The size of datasets and demands of downstream workflows are growing rapidly, making robust and scalable software essential. This work presents a major overhaul of the NequIP framework focusing on multi-node parallelism, computational performance, and extensibility. The redesigned framework supports distributed training on large datasets and removes barriers preventing full utilization of the PyTorch 2.0 compiler at train time. We demonstrate this acceleration in a case study by training Allegro models on the SPICE 2 dataset of organic molecular systems. For inference, we introduce the first end-to-end infrastructure that uses the PyTorch Ahead-of-Time Inductor compiler for machine learning interatomic potentials. Additionally, we implement a custom kernel for the Allegro model's most expensive operation, the tensor product. Together, these advancements speed up molecular dynamics calculations on system sizes of practical relevance by up to a factor of 18.
A practical guide to machine learning interatomic potentials -- Status and future
Mar 12, 2025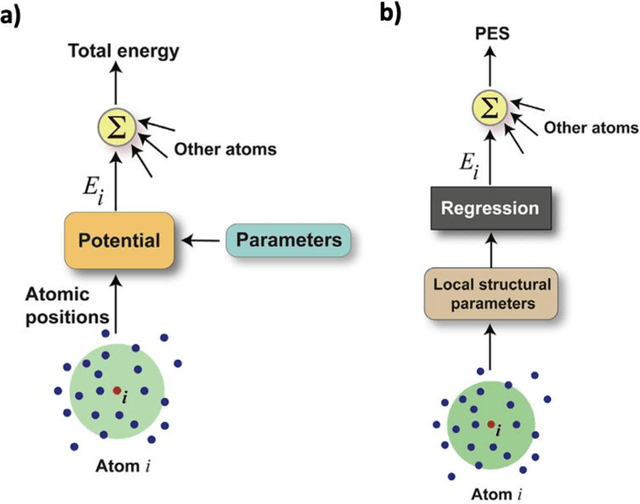

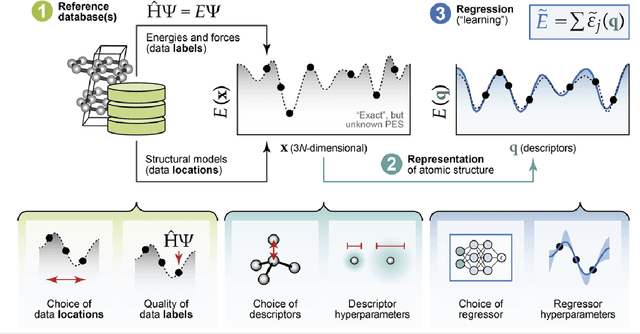
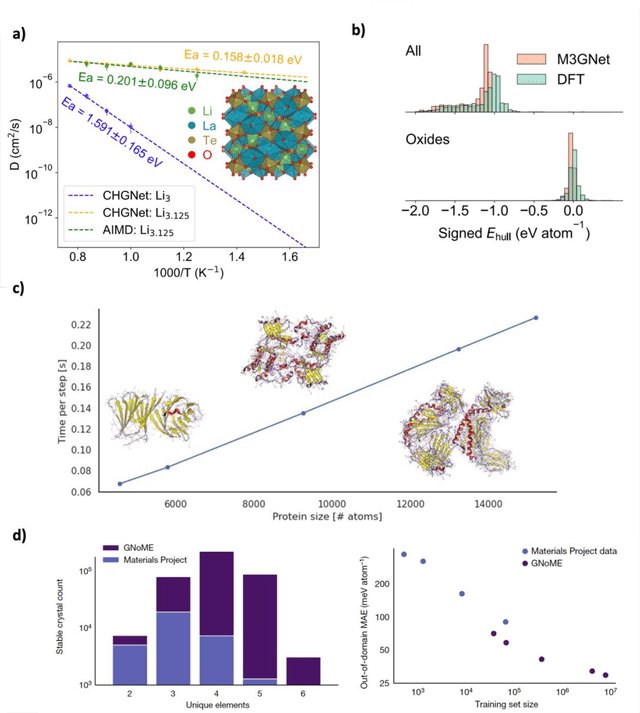
The rapid development and large body of literature on machine learning interatomic potentials (MLIPs) can make it difficult to know how to proceed for researchers who are not experts but wish to use these tools. The spirit of this review is to help such researchers by serving as a practical, accessible guide to the state-of-the-art in MLIPs. This review paper covers a broad range of topics related to MLIPs, including (i) central aspects of how and why MLIPs are enablers of many exciting advancements in molecular modeling, (ii) the main underpinnings of different types of MLIPs, including their basic structure and formalism, (iii) the potentially transformative impact of universal MLIPs for both organic and inorganic systems, including an overview of the most recent advances, capabilities, downsides, and potential applications of this nascent class of MLIPs, (iv) a practical guide for estimating and understanding the execution speed of MLIPs, including guidance for users based on hardware availability, type of MLIP used, and prospective simulation size and time, (v) a manual for what MLIP a user should choose for a given application by considering hardware resources, speed requirements, energy and force accuracy requirements, as well as guidance for choosing pre-trained potentials or fitting a new potential from scratch, (vi) discussion around MLIP infrastructure, including sources of training data, pre-trained potentials, and hardware resources for training, (vii) summary of some key limitations of present MLIPs and current approaches to mitigate such limitations, including methods of including long-range interactions, handling magnetic systems, and treatment of excited states, and finally (viii) we finish with some more speculative thoughts on what the future holds for the development and application of MLIPs over the next 3-10+ years.
Phase discovery with active learning: Application to structural phase transitions in equiatomic NiTi
Jan 10, 2024Nickel titanium (NiTi) is a protypical shape-memory alloy used in a range of biomedical and engineering devices, but direct molecular dynamics simulations of the martensitic B19' -> B2 phase transition driving its shape-memory behavior are rare and have relied on classical force fields with limited accuracy. Here, we train four machine-learned force fields for equiatomic NiTi based on the LDA, PBE, PBEsol, and SCAN DFT functionals. The models are trained on the fly during NPT molecular dynamics, with DFT calculations and model updates performed automatically whenever the uncertainty of a local energy prediction exceeds a chosen threshold. The models achieve accuracies of 1-2 meV/atom during training and are shown to closely track DFT predictions of B2 and B19' elastic constants and phonon frequencies. Surprisingly, in large-scale molecular dynamics simulations, only the SCAN model predicts a reversible B19' -> B2 phase transition, with the LDA, PBE, and PBEsol models predicting a reversible transition to a previously uncharacterized low-volume phase, which we hypothesize to be a new stable high-pressure phase. We examine the structure of the new phase and estimate its stability on the temperature-pressure phase diagram. This work establishes an automated active learning protocol for studying displacive transformations, reveals important differences between DFT functionals that can only be detected in large-scale simulations, provides an accurate force field for NiTi, and identifies a new phase.
Learning Interatomic Potentials at Multiple Scales
Oct 20, 2023The need to use a short time step is a key limit on the speed of molecular dynamics (MD) simulations. Simulations governed by classical potentials are often accelerated by using a multiple-time-step (MTS) integrator that evaluates certain potential energy terms that vary more slowly than others less frequently. This approach is enabled by the simple but limiting analytic forms of classical potentials. Machine learning interatomic potentials (MLIPs), in particular recent equivariant neural networks, are much more broadly applicable than classical potentials and can faithfully reproduce the expensive but accurate reference electronic structure calculations used to train them. They still, however, require the use of a single short time step, as they lack the inherent term-by-term scale separation of classical potentials. This work introduces a method to learn a scale separation in complex interatomic interactions by co-training two MLIPs. Initially, a small and efficient model is trained to reproduce short-time-scale interactions. Subsequently, a large and expressive model is trained jointly to capture the remaining interactions not captured by the small model. When running MD, the MTS integrator then evaluates the smaller model for every time step and the larger model less frequently, accelerating simulation. Compared to a conventionally trained MLIP, our approach can achieve a significant speedup (~3x in our experiments) without a loss of accuracy on the potential energy or simulation-derived quantities.
Scaling the leading accuracy of deep equivariant models to biomolecular simulations of realistic size
Apr 20, 2023
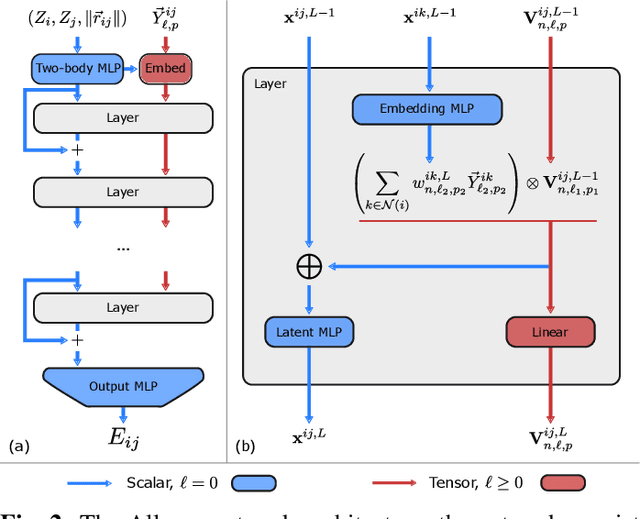
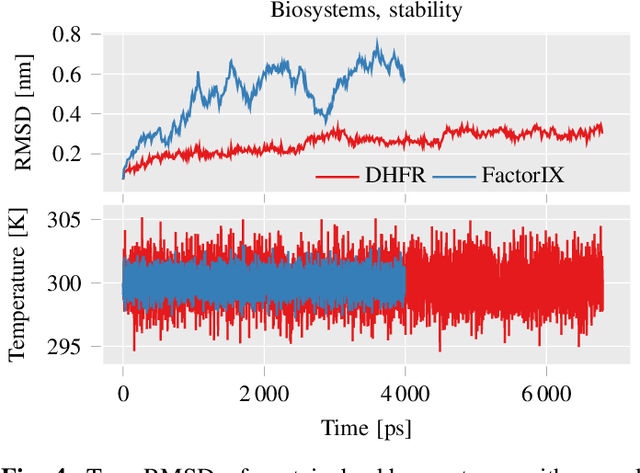
This work brings the leading accuracy, sample efficiency, and robustness of deep equivariant neural networks to the extreme computational scale. This is achieved through a combination of innovative model architecture, massive parallelization, and models and implementations optimized for efficient GPU utilization. The resulting Allegro architecture bridges the accuracy-speed tradeoff of atomistic simulations and enables description of dynamics in structures of unprecedented complexity at quantum fidelity. To illustrate the scalability of Allegro, we perform nanoseconds-long stable simulations of protein dynamics and scale up to a 44-million atom structure of a complete, all-atom, explicitly solvated HIV capsid on the Perlmutter supercomputer. We demonstrate excellent strong scaling up to 100 million atoms and 70% weak scaling to 5120 A100 GPUs.
Learning Local Equivariant Representations for Large-Scale Atomistic Dynamics
Apr 11, 2022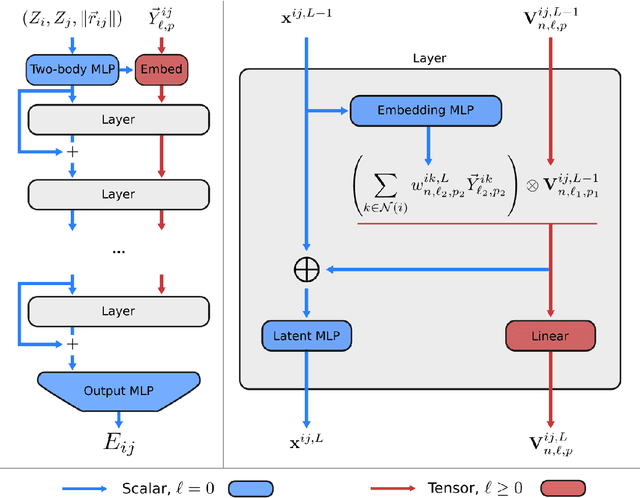
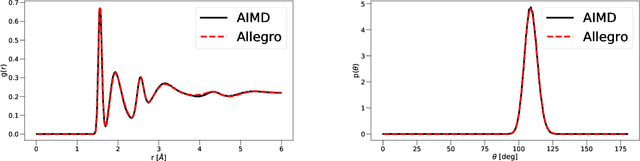
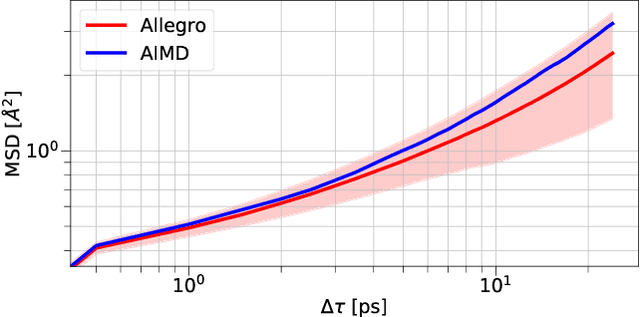
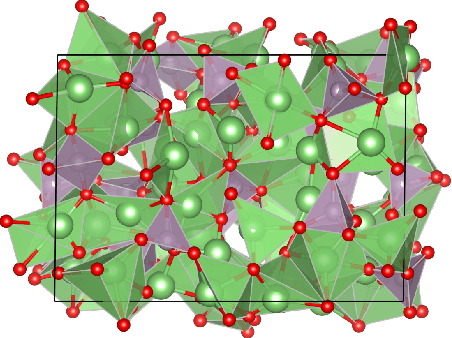
A simultaneously accurate and computationally efficient parametrization of the energy and atomic forces of molecules and materials is a long-standing goal in the natural sciences. In pursuit of this goal, neural message passing has lead to a paradigm shift by describing many-body correlations of atoms through iteratively passing messages along an atomistic graph. This propagation of information, however, makes parallel computation difficult and limits the length scales that can be studied. Strictly local descriptor-based methods, on the other hand, can scale to large systems but do not currently match the high accuracy observed with message passing approaches. This work introduces Allegro, a strictly local equivariant deep learning interatomic potential that simultaneously exhibits excellent accuracy and scalability of parallel computation. Allegro learns many-body functions of atomic coordinates using a series of tensor products of learned equivariant representations, but without relying on message passing. Allegro obtains improvements over state-of-the-art methods on the QM9 and revised MD-17 data sets. A single tensor product layer is shown to outperform existing deep message passing neural networks and transformers on the QM9 benchmark. Furthermore, Allegro displays remarkable generalization to out-of-distribution data. Molecular dynamics simulations based on Allegro recover structural and kinetic properties of an amorphous phosphate electrolyte in excellent agreement with first principles calculations. Finally, we demonstrate the parallel scaling of Allegro with a dynamics simulation of 100 million atoms.