 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImplicit Neural Representations for Chemical Reaction Paths
Feb 20, 2025We show that neural networks can be optimized to represent minimum energy paths as continuous functions, offering a flexible alternative to discrete path-search methods like Nudged Elastic Band (NEB). Our approach parameterizes reaction paths with a network trained on a loss function that discards tangential energy gradients and enables instant estimation of the transition state. We first validate the method on two-dimensional potentials and then demonstrate its advantages over NEB on challenging atomistic systems where (i) poor initial guesses yield unphysical paths, (ii) multiple competing paths exist, or (iii) the reaction follows a complex multi-step mechanism. Results highlight the versatility of the method -- for instance, a simple adjustment to the sampling strategy during optimization can help escape local-minimum solutions. Finally, in a low-dimensional setting, we demonstrate that a single neural network can learn from existing paths and generalize to unseen systems, showing promise for a universal reaction path representation.
BoostMD: Accelerating molecular sampling by leveraging ML force field features from previous time-steps
Dec 21, 2024Simulating atomic-scale processes, such as protein dynamics and catalytic reactions, is crucial for advancements in biology, chemistry, and materials science. Machine learning force fields (MLFFs) have emerged as powerful tools that achieve near quantum mechanical accuracy, with promising generalization capabilities. However, their practical use is often limited by long inference times compared to classical force fields, especially when running extensive molecular dynamics (MD) simulations required for many biological applications. In this study, we introduce BoostMD, a surrogate model architecture designed to accelerate MD simulations. BoostMD leverages node features computed at previous time steps to predict energies and forces based on positional changes. This approach reduces the complexity of the learning task, allowing BoostMD to be both smaller and significantly faster than conventional MLFFs. During simulations, the computationally intensive reference MLFF is evaluated only every $N$ steps, while the lightweight BoostMD model handles the intermediate steps at a fraction of the computational cost. Our experiments demonstrate that BoostMD achieves an eight-fold speedup compared to the reference model and generalizes to unseen dipeptides. Furthermore, we find that BoostMD accurately samples the ground-truth Boltzmann distribution when running molecular dynamics. By combining efficient feature reuse with a streamlined architecture, BoostMD offers a robust solution for conducting large-scale, long-timescale molecular simulations, making high-accuracy ML-driven modeling more accessible and practical.
Equivariant Matrix Function Neural Networks
Oct 16, 2023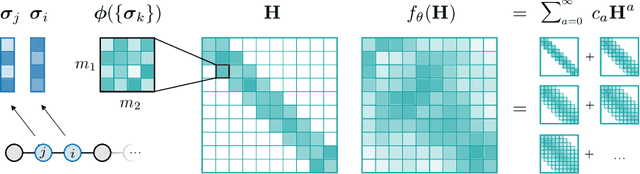
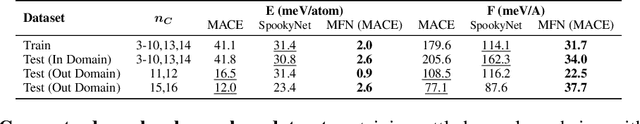
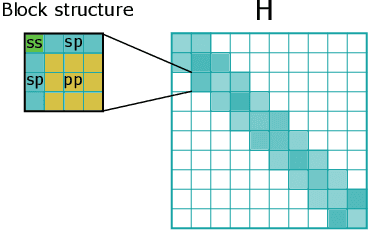

Graph Neural Networks (GNNs), especially message-passing neural networks (MPNNs), have emerged as powerful architectures for learning on graphs in diverse applications. However, MPNNs face challenges when modeling non-local interactions in systems such as large conjugated molecules, metals, or amorphous materials. Although Spectral GNNs and traditional neural networks such as recurrent neural networks and transformers mitigate these challenges, they often lack extensivity, adaptability, generalizability, computational efficiency, or fail to capture detailed structural relationships or symmetries in the data. To address these concerns, we introduce Matrix Function Neural Networks (MFNs), a novel architecture that parameterizes non-local interactions through analytic matrix equivariant functions. Employing resolvent expansions offers a straightforward implementation and the potential for linear scaling with system size. The MFN architecture achieves state-of-the-art performance in standard graph benchmarks, such as the ZINC and TU datasets, and is able to capture intricate non-local interactions in quantum systems, paving the way to new state-of-the-art force fields.