 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHyperactive Learning (HAL) for Data-Driven Interatomic Potentials
Paper and Code
Oct 09, 2022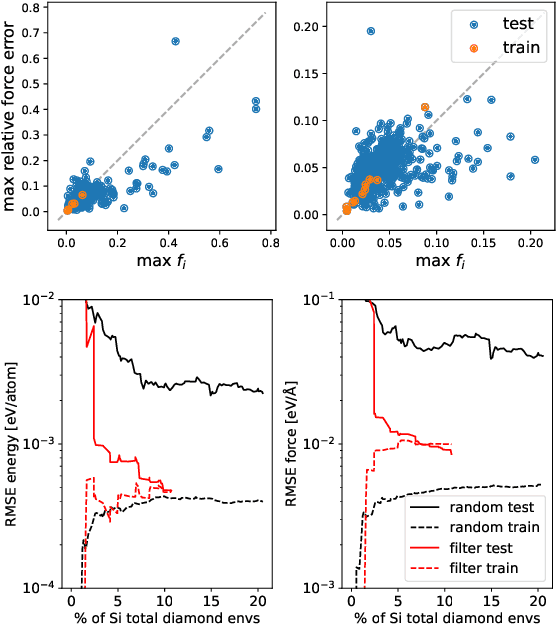
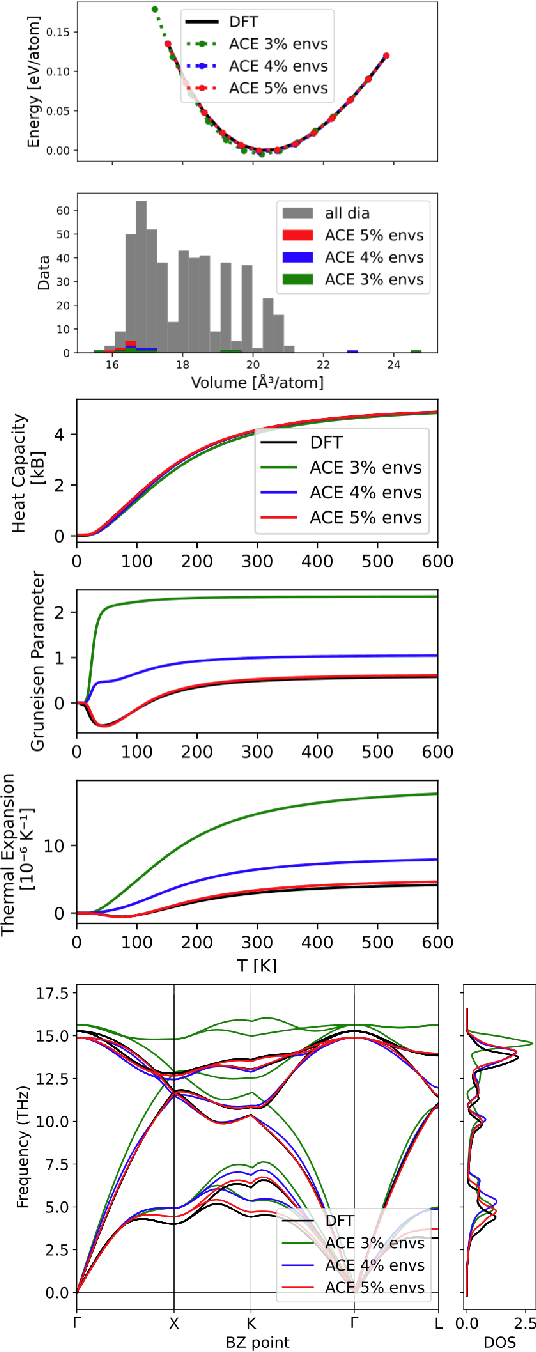
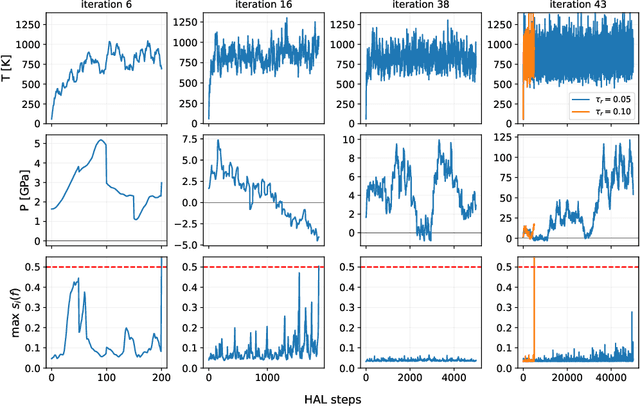
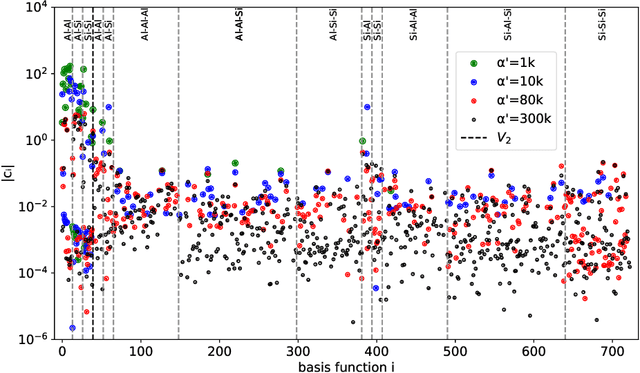
Data-driven interatomic potentials have emerged as a powerful class of surrogate models for ab initio potential energy surfaces that are able to reliably predict macroscopic properties with experimental accuracy. In generating accurate and transferable potentials the most time-consuming and arguably most important task is generating the training set, which still requires significant expert user input. To accelerate this process, this work presents hyperactive learning (HAL), a framework for formulating an accelerated sampling algorithm specifically for the task of training database generation. The overarching idea is to start from a physically motivated sampler (e.g., molecular dynamics) and a biasing term that drives the system towards high uncertainty and thus to unseen training configurations. Building on this framework, general protocols for building training databases for alloys and polymers leveraging the HAL framework will be presented. For alloys, fast (< 100 microsecond/atom/cpu-core) ACE potentials for AlSi10 are created that able to predict the melting temperature with good accuracy by fitting to a minimal HAL-generated database containing 88 configurations (32 atoms each) in 17 seconds using 8 cpu threads. For polymers, a HAL database is built using ACE able to determine the density of a long polyethylene glycol (PEG) polymer formed of 200 monomer units with experimental accuracy by only fitting to small isolated PEG polymers with sizes ranging from 2 to 32.