 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCrystalGRW: Generative Modeling of Crystal Structures with Targeted Properties via Geodesic Random Walks
Jan 15, 2025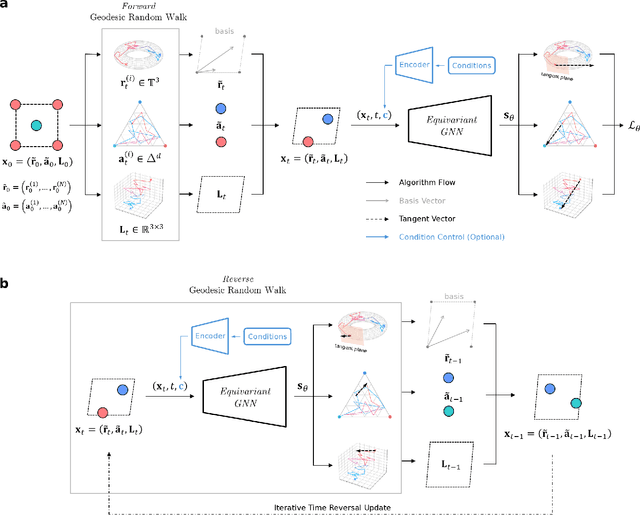
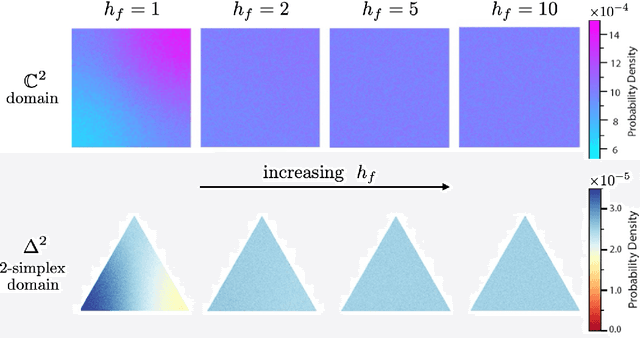
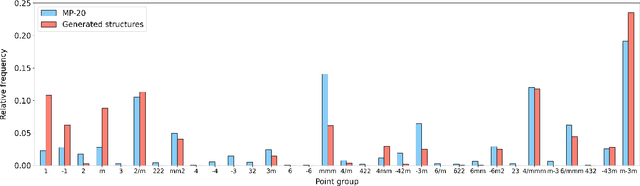
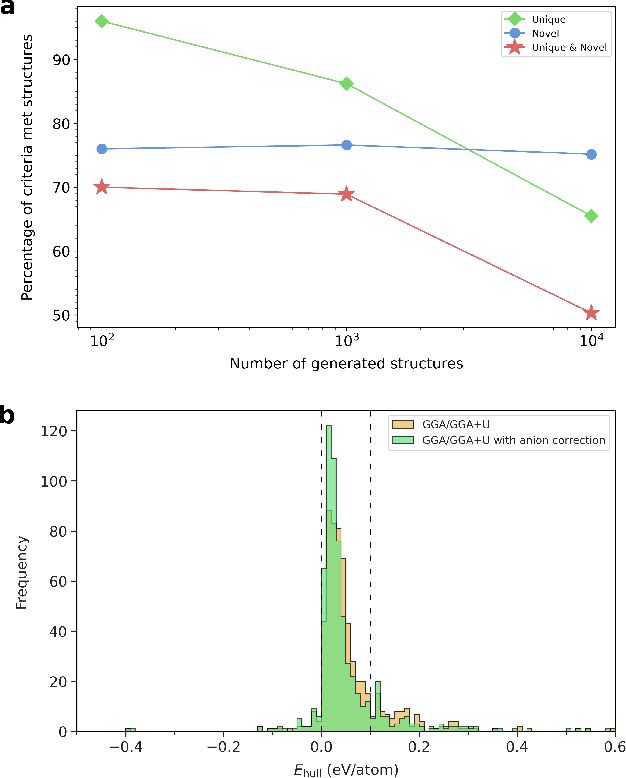
Determining whether a candidate crystalline material is thermodynamically stable depends on identifying its true ground-state structure, a central challenge in computational materials science. We introduce CrystalGRW, a diffusion-based generative model on Riemannian manifolds that proposes novel crystal configurations and can predict stable phases validated by density functional theory. The crystal properties, such as fractional coordinates, atomic types, and lattice matrices, are represented on suitable Riemannian manifolds, ensuring that new predictions generated through the diffusion process preserve the periodicity of crystal structures. We incorporate an equivariant graph neural network to also account for rotational and translational symmetries during the generation process. CrystalGRW demonstrates the ability to generate realistic crystal structures that are close to their ground states with accuracy comparable to existing models, while also enabling conditional control, such as specifying a desired crystallographic point group. These features help accelerate materials discovery and inverse design by offering stable, symmetry-consistent crystal candidates for experimental validation.
Accurate Crystal Structure Prediction of New 2D Hybrid Organic Inorganic Perovskites
Mar 11, 2024Low dimensional hybrid organic-inorganic perovskites (HOIPs) represent a promising class of electronically active materials for both light absorption and emission. The design space of HOIPs is extremely large, since a diverse space of organic cations can be combined with different inorganic frameworks. This immense design space allows for tunable electronic and mechanical properties, but also necessitates the development of new tools for in silico high throughput analysis of candidate structures. In this work, we present an accurate, efficient, transferable and widely applicable machine learning interatomic potential (MLIP) for predicting the structure of new 2D HOIPs. Using the MACE architecture, an MLIP is trained on 86 diverse experimentally reported HOIP structures. The model is tested on 73 unseen perovskite compositions, and achieves chemical accuracy with respect to the reference electronic structure method. Our model is then combined with a simple random structure search algorithm to predict the structure of hypothetical HOIPs given only the proposed composition. Success is demonstrated by correctly and reliably recovering the crystal structure of a set of experimentally known 2D perovskites. Such a random structure search is impossible with ab initio methods due to the associated computational cost, but is relatively inexpensive with the MACE potential. Finally, the procedure is used to predict the structure formed by a new organic cation with no previously known corresponding perovskite. Laboratory synthesis of the new hybrid perovskite confirms the accuracy of our prediction. This capability, applied at scale, enables efficient screening of thousands of combinations of organic cations and inorganic layers.
From structure mining to unsupervised exploration of atomic octahedral networks
Jun 21, 2023



Networks of atom-centered coordination octahedra commonly occur in inorganic and hybrid solid-state materials. Characterizing their spatial arrangements and characteristics is crucial for relating structures to properties for many materials families. The traditional method using case-by-case inspection becomes prohibitive for discovering trends and similarities in large datasets. Here, we operationalize chemical intuition to automate the geometric parsing, quantification, and classification of coordination octahedral networks. We find axis-resolved tilting trends in ABO$_{3}$ perovskite polymorphs, which assist in detecting oxidation state changes. Moreover, we develop a scale-invariant encoding scheme to represent these networks, which, combined with human-assisted unsupervised machine learning, allows us to taxonomize the inorganic framework polytypes in hybrid iodoplumbates (A$_x$Pb$_y$I$_z$). Consequently, we uncover a violation of Pauling's third rule and the design principles underpinning their topological diversity. Our results offer a glimpse into the vast design space of atomic octahedral networks and inform high-throughput, targeted screening of specific structure types.