 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Generative Model for Molecular Distance Geometry
Paper and Code
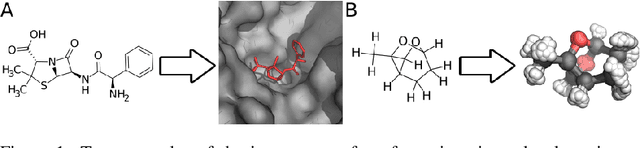


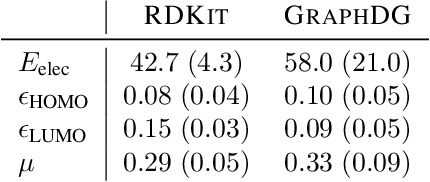
Computing equilibrium states for many-body systems, such as molecules, is a long-standing challenge. In the absence of methods for generating statistically independent samples, great computational effort is invested in simulating these systems using, for example, Markov chain Monte Carlo. We present a probabilistic model that generates such samples for molecules from their graph representations. Our model learns a low-dimensional manifold that preserves the geometry of local atomic neighborhoods through a principled learning representation that is based on Euclidean distance geometry. We create a new dataset for molecular conformation generation with which we show experimentally that our generative model achieves state-of-the-art accuracy. Finally, we show how to use our model as a proposal distribution in an importance sampling scheme to compute molecular properties.