 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeClosed-Form Test Functions for Biophysical Sequence Optimization Algorithms
Jun 28, 2024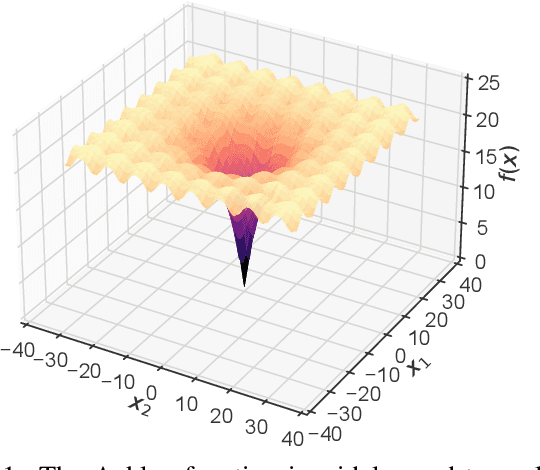

There is a growing body of work seeking to replicate the success of machine learning (ML) on domains like computer vision (CV) and natural language processing (NLP) to applications involving biophysical data. One of the key ingredients of prior successes in CV and NLP was the broad acceptance of difficult benchmarks that distilled key subproblems into approachable tasks that any junior researcher could investigate, but good benchmarks for biophysical domains are rare. This scarcity is partially due to a narrow focus on benchmarks which simulate biophysical data; we propose instead to carefully abstract biophysical problems into simpler ones with key geometric similarities. In particular we propose a new class of closed-form test functions for biophysical sequence optimization, which we call Ehrlich functions. We provide empirical results demonstrating these functions are interesting objects of study and can be non-trivial to solve with a standard genetic optimization baseline.
MoleCLUEs: Optimizing Molecular Conformers by Minimization of Differentiable Uncertainty
Jun 20, 2023



Structure-based models in the molecular sciences can be highly sensitive to input geometries and give predictions with large variance under subtle coordinate perturbations. We present an approach to mitigate this failure mode by generating conformations that explicitly minimize uncertainty in a predictive model. To achieve this, we compute differentiable estimates of aleatoric \textit{and} epistemic uncertainties directly from learned embeddings. We then train an optimizer that iteratively samples embeddings to reduce these uncertainties according to their gradients. As our predictive model is constructed as a variational autoencoder, the new embeddings can be decoded to their corresponding inputs, which we call \textit{MoleCLUEs}, or (molecular) counterfactual latent uncertainty explanations \citep{antoran2020getting}. We provide results of our algorithm for the task of predicting drug properties with maximum confidence as well as analysis of the differentiable structure simulations.
SupSiam: Non-contrastive Auxiliary Loss for Learning from Molecular Conformers
Feb 15, 2023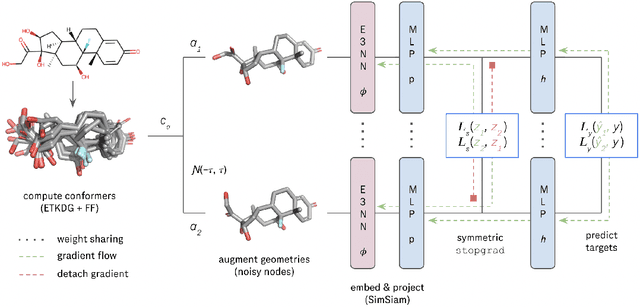


We investigate Siamese networks for learning related embeddings for augmented samples of molecular conformers. We find that a non-contrastive (positive-pair only) auxiliary task aids in supervised training of Euclidean neural networks (E3NNs) and increases manifold smoothness (MS) around point-cloud geometries. We demonstrate this property for multiple drug-activity prediction tasks while maintaining relevant performance metrics, and propose an extension of MS to probabilistic and regression settings. We provide an analysis of representation collapse, finding substantial effects of task-weighting, latent dimension, and regularization. We expect the presented protocol to aid in the development of reliable E3NNs from molecular conformers, even for small-data drug discovery programs.
PropertyDAG: Multi-objective Bayesian optimization of partially ordered, mixed-variable properties for biological sequence design
Oct 08, 2022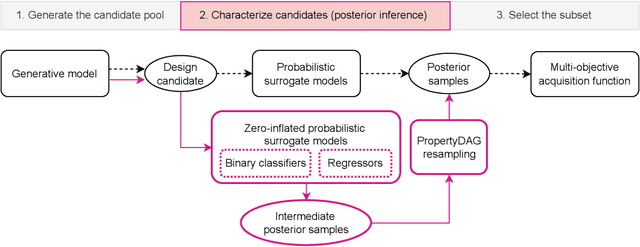
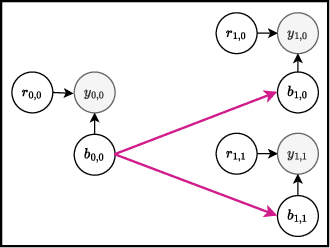
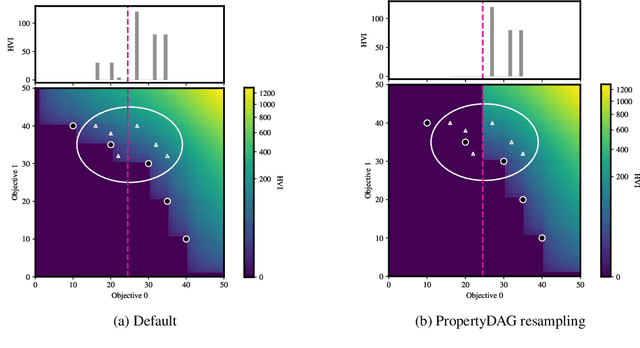
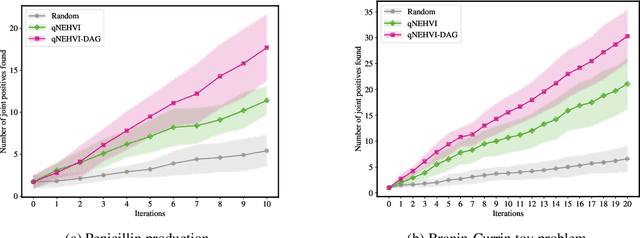
Bayesian optimization offers a sample-efficient framework for navigating the exploration-exploitation trade-off in the vast design space of biological sequences. Whereas it is possible to optimize the various properties of interest jointly using a multi-objective acquisition function, such as the expected hypervolume improvement (EHVI), this approach does not account for objectives with a hierarchical dependency structure. We consider a common use case where some regions of the Pareto frontier are prioritized over others according to a specified $\textit{partial ordering}$ in the objectives. For instance, when designing antibodies, we would like to maximize the binding affinity to a target antigen only if it can be expressed in live cell culture -- modeling the experimental dependency in which affinity can only be measured for antibodies that can be expressed and thus produced in viable quantities. In general, we may want to confer a partial ordering to the properties such that each property is optimized conditioned on its parent properties satisfying some feasibility condition. To this end, we present PropertyDAG, a framework that operates on top of the traditional multi-objective BO to impose this desired ordering on the objectives, e.g. expression $\rightarrow$ affinity. We demonstrate its performance over multiple simulated active learning iterations on a penicillin production task, toy numerical problem, and a real-world antibody design task.
Multi-segment preserving sampling for deep manifold sampler
May 09, 2022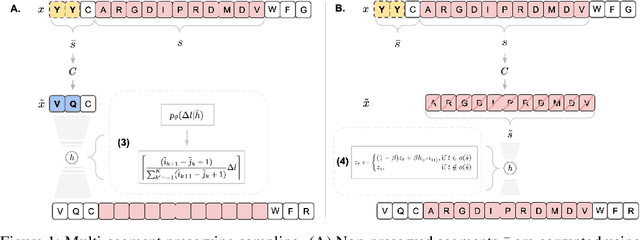

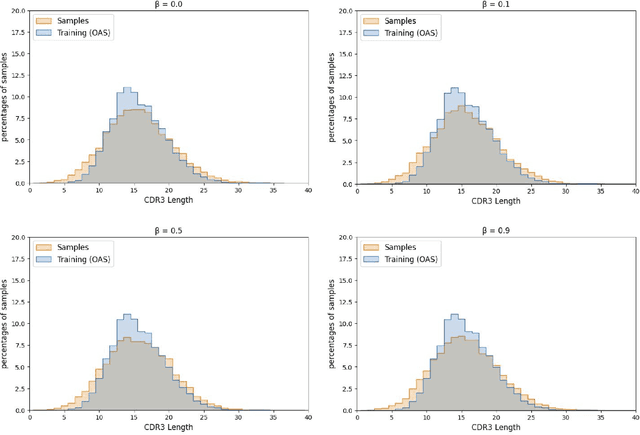
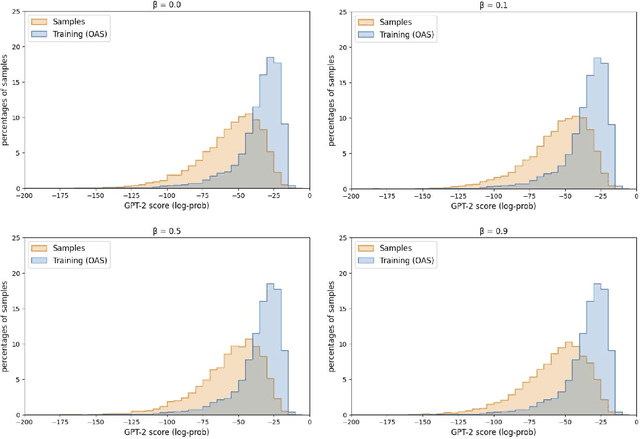
Deep generative modeling for biological sequences presents a unique challenge in reconciling the bias-variance trade-off between explicit biological insight and model flexibility. The deep manifold sampler was recently proposed as a means to iteratively sample variable-length protein sequences by exploiting the gradients from a function predictor. We introduce an alternative approach to this guided sampling procedure, multi-segment preserving sampling, that enables the direct inclusion of domain-specific knowledge by designating preserved and non-preserved segments along the input sequence, thereby restricting variation to only select regions. We present its effectiveness in the context of antibody design by training two models: a deep manifold sampler and a GPT-2 language model on nearly six million heavy chain sequences annotated with the IGHV1-18 gene. During sampling, we restrict variation to only the complementarity-determining region 3 (CDR3) of the input. We obtain log probability scores from a GPT-2 model for each sampled CDR3 and demonstrate that multi-segment preserving sampling generates reasonable designs while maintaining the desired, preserved regions.