 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative Active Learning for the Search of Small-molecule Protein Binders
May 02, 2024



Despite substantial progress in machine learning for scientific discovery in recent years, truly de novo design of small molecules which exhibit a property of interest remains a significant challenge. We introduce LambdaZero, a generative active learning approach to search for synthesizable molecules. Powered by deep reinforcement learning, LambdaZero learns to search over the vast space of molecules to discover candidates with a desired property. We apply LambdaZero with molecular docking to design novel small molecules that inhibit the enzyme soluble Epoxide Hydrolase 2 (sEH), while enforcing constraints on synthesizability and drug-likeliness. LambdaZero provides an exponential speedup in terms of the number of calls to the expensive molecular docking oracle, and LambdaZero de novo designed molecules reach docking scores that would otherwise require the virtual screening of a hundred billion molecules. Importantly, LambdaZero discovers novel scaffolds of synthesizable, drug-like inhibitors for sEH. In in vitro experimental validation, a series of ligands from a generated quinazoline-based scaffold were synthesized, and the lead inhibitor N-(4,6-di(pyrrolidin-1-yl)quinazolin-2-yl)-N-methylbenzamide (UM0152893) displayed sub-micromolar enzyme inhibition of sEH.
PDB-Struct: A Comprehensive Benchmark for Structure-based Protein Design
Nov 30, 2023



Structure-based protein design has attracted increasing interest, with numerous methods being introduced in recent years. However, a universally accepted method for evaluation has not been established, since the wet-lab validation can be overly time-consuming for the development of new algorithms, and the $\textit{in silico}$ validation with recovery and perplexity metrics is efficient but may not precisely reflect true foldability. To address this gap, we introduce two novel metrics: refoldability-based metric, which leverages high-accuracy protein structure prediction models as a proxy for wet lab experiments, and stability-based metric, which assesses whether models can assign high likelihoods to experimentally stable proteins. We curate datasets from high-quality CATH protein data, high-throughput $\textit{de novo}$ designed proteins, and mega-scale experimental mutagenesis experiments, and in doing so, present the $\textbf{PDB-Struct}$ benchmark that evaluates both recent and previously uncompared protein design methods. Experimental results indicate that ByProt, ProteinMPNN, and ESM-IF perform exceptionally well on our benchmark, while ESM-Design and AF-Design fall short on the refoldability metric. We also show that while some methods exhibit high sequence recovery, they do not perform as well on our new benchmark. Our proposed benchmark paves the way for a fair and comprehensive evaluation of protein design methods in the future. Code is available at https://github.com/WANG-CR/PDB-Struct.
DiffPack: A Torsional Diffusion Model for Autoregressive Protein Side-Chain Packing
Jun 01, 2023Proteins play a critical role in carrying out biological functions, and their 3D structures are essential in determining their functions. Accurately predicting the conformation of protein side-chains given their backbones is important for applications in protein structure prediction, design and protein-protein interactions. Traditional methods are computationally intensive and have limited accuracy, while existing machine learning methods treat the problem as a regression task and overlook the restrictions imposed by the constant covalent bond lengths and angles. In this work, we present DiffPack, a torsional diffusion model that learns the joint distribution of side-chain torsional angles, the only degrees of freedom in side-chain packing, by diffusing and denoising on the torsional space. To avoid issues arising from simultaneous perturbation of all four torsional angles, we propose autoregressively generating the four torsional angles from \c{hi}1 to \c{hi}4 and training diffusion models for each torsional angle. We evaluate the method on several benchmarks for protein side-chain packing and show that our method achieves improvements of 11.9% and 13.5% in angle accuracy on CASP13 and CASP14, respectively, with a significantly smaller model size (60x fewer parameters). Additionally, we show the effectiveness of our method in enhancing side-chain predictions in the AlphaFold2 model. Code will be available upon the accept.
Accelerating Barnes-Hut t-SNE Algorithm by Efficient Parallelization on Multi-Core CPUs
Dec 22, 2022



t-SNE remains one of the most popular embedding techniques for visualizing high-dimensional data. Most standard packages of t-SNE, such as scikit-learn, use the Barnes-Hut t-SNE (BH t-SNE) algorithm for large datasets. However, existing CPU implementations of this algorithm are inefficient. In this work, we accelerate the BH t-SNE on CPUs via cache optimizations, SIMD, parallelizing sequential steps, and improving parallelization of multithreaded steps. Our implementation (Acc-t-SNE) is up to 261x and 4x faster than scikit-learn and the state-of-the-art BH t-SNE implementation from daal4py, respectively, on a 32-core Intel(R) Icelake cloud instance.
DistGNN-MB: Distributed Large-Scale Graph Neural Network Training on x86 via Minibatch Sampling
Nov 11, 2022

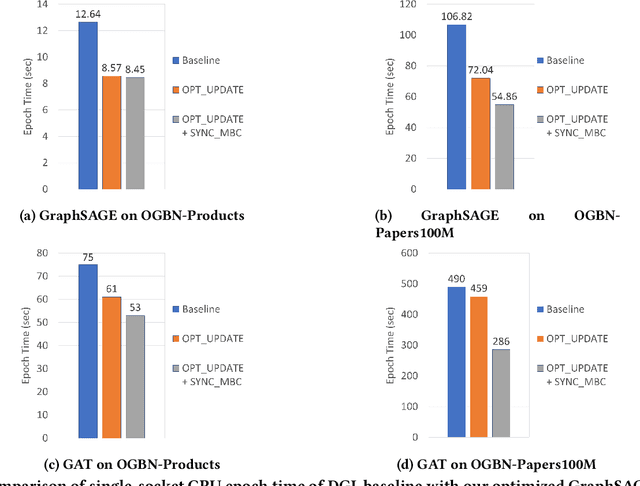
Training Graph Neural Networks, on graphs containing billions of vertices and edges, at scale using minibatch sampling poses a key challenge: strong-scaling graphs and training examples results in lower compute and higher communication volume and potential performance loss. DistGNN-MB employs a novel Historical Embedding Cache combined with compute-communication overlap to address this challenge. On a 32-node (64-socket) cluster of $3^{rd}$ generation Intel Xeon Scalable Processors with 36 cores per socket, DistGNN-MB trains 3-layer GraphSAGE and GAT models on OGBN-Papers100M to convergence with epoch times of 2 seconds and 4.9 seconds, respectively, on 32 compute nodes. At this scale, DistGNN-MB trains GraphSAGE 5.2x faster than the widely-used DistDGL. DistGNN-MB trains GraphSAGE and GAT 10x and 17.2x faster, respectively, as compute nodes scale from 2 to 32.
DistGNN: Scalable Distributed Training for Large-Scale Graph Neural Networks
Apr 16, 2021
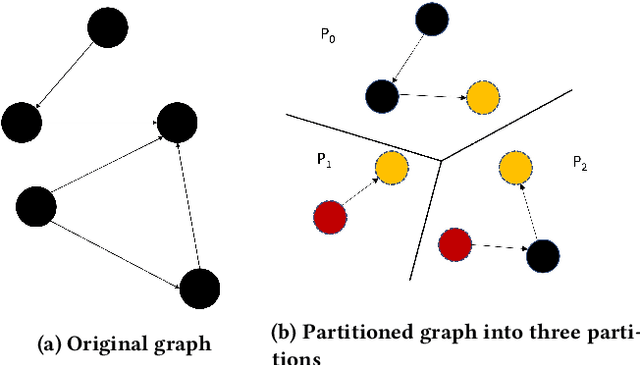
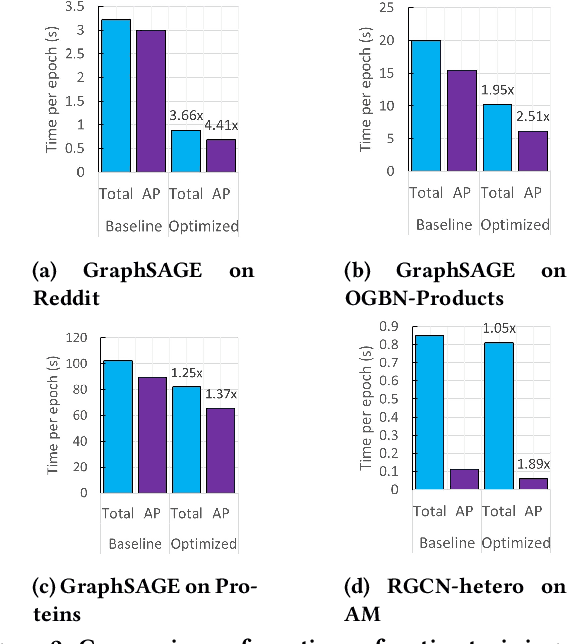
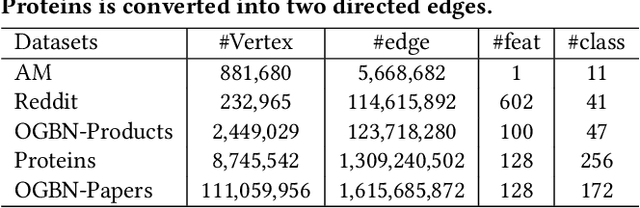
Full-batch training on Graph Neural Networks (GNN) to learn the structure of large graphs is a critical problem that needs to scale to hundreds of compute nodes to be feasible. It is challenging due to large memory capacity and bandwidth requirements on a single compute node and high communication volumes across multiple nodes. In this paper, we present DistGNN that optimizes the well-known Deep Graph Library (DGL) for full-batch training on CPU clusters via an efficient shared memory implementation, communication reduction using a minimum vertex-cut graph partitioning algorithm and communication avoidance using a family of delayed-update algorithms. Our results on four common GNN benchmark datasets: Reddit, OGB-Products, OGB-Papers and Proteins, show up to 3.7x speed-up using a single CPU socket and up to 97x speed-up using 128 CPU sockets, respectively, over baseline DGL implementations running on a single CPU socket
Efficient and Generic 1D Dilated Convolution Layer for Deep Learning
Apr 16, 2021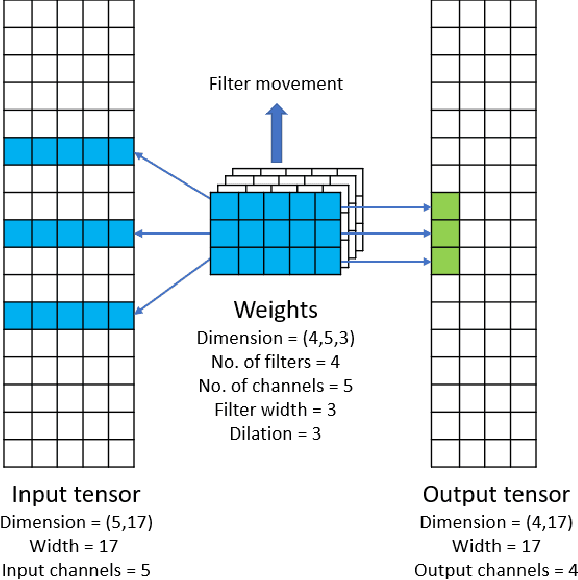
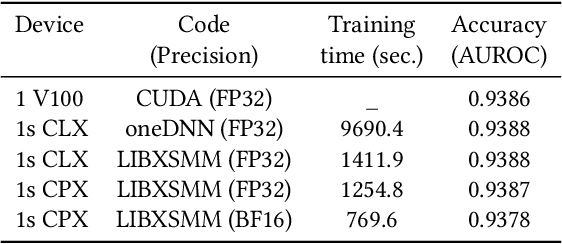
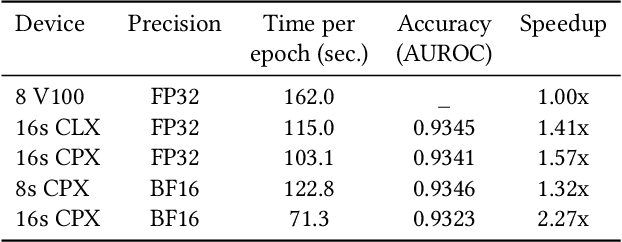
Convolutional neural networks (CNNs) have found many applications in tasks involving two-dimensional (2D) data, such as image classification and image processing. Therefore, 2D convolution layers have been heavily optimized on CPUs and GPUs. However, in many applications - for example genomics and speech recognition, the data can be one-dimensional (1D). Such applications can benefit from optimized 1D convolution layers. In this work, we introduce our efficient implementation of a generic 1D convolution layer covering a wide range of parameters. It is optimized for x86 CPU architectures, in particular, for architectures containing Intel AVX-512 and AVX-512 BFloat16 instructions. We use the LIBXSMM library's batch-reduce General Matrix Multiplication (BRGEMM) kernel for FP32 and BFloat16 precision. We demonstrate that our implementation can achieve up to 80% efficiency on Intel Xeon Cascade Lake and Cooper Lake CPUs. Additionally, we show the generalization capability of our BRGEMM based approach by achieving high efficiency across a range of parameters. We consistently achieve higher efficiency than the 1D convolution layer with Intel oneDNN library backend for varying input tensor widths, filter widths, number of channels, filters, and dilation parameters. Finally, we demonstrate the performance of our optimized 1D convolution layer by utilizing it in the end-to-end neural network training with real genomics datasets and achieve up to 6.86x speedup over the oneDNN library-based implementation on Cascade Lake CPUs. We also demonstrate the scaling with 16 sockets of Cascade/Cooper Lake CPUs and achieve significant speedup over eight V100 GPUs using a similar power envelop. In the end-to-end training, we get a speedup of 1.41x on Cascade Lake with FP32, 1.57x on Cooper Lake with FP32, and 2.27x on Cooper Lake with BFloat16 over eight V100 GPUs with FP32.
Tensor Processing Primitives: A Programming Abstraction for Efficiency and Portability in Deep Learning Workloads
Apr 14, 2021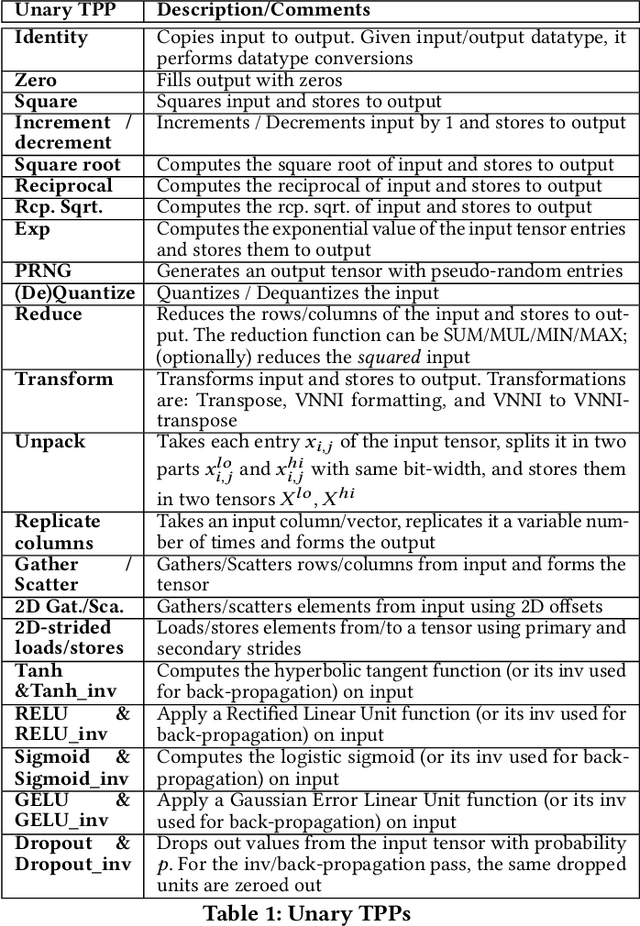
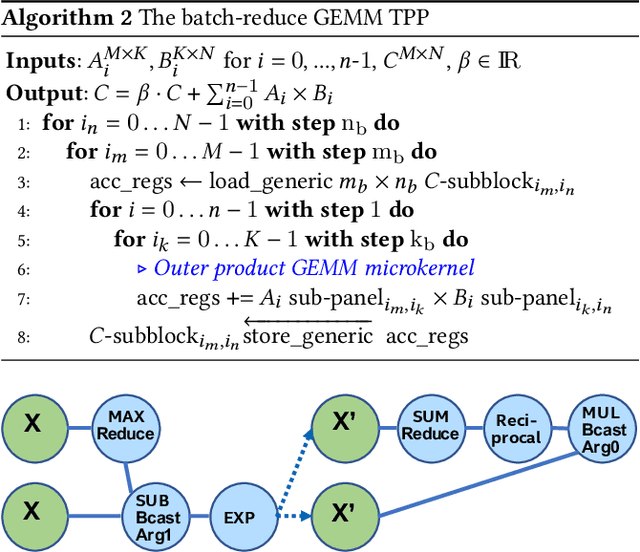
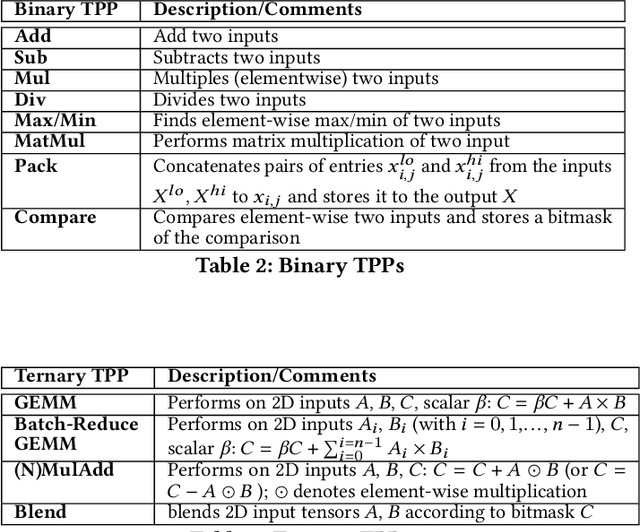
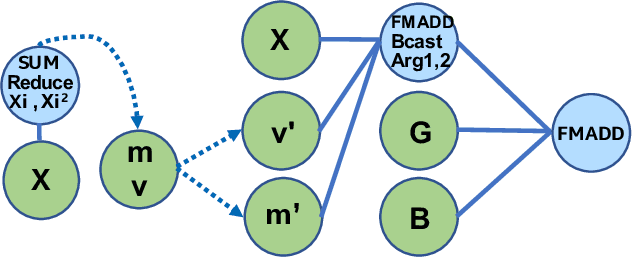
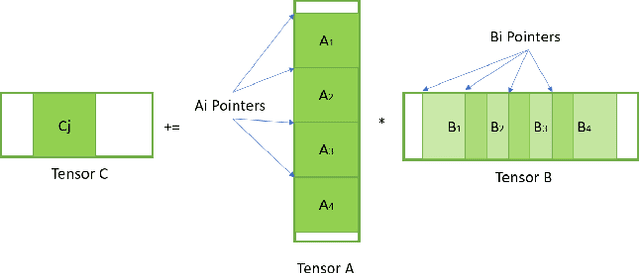
During the past decade, novel Deep Learning (DL) algorithms/workloads and hardware have been developed to tackle a wide range of problems. Despite the advances in workload/hardware ecosystems, the programming methodology of DL-systems is stagnant. DL-workloads leverage either highly-optimized, yet platform-specific and inflexible kernels from DL-libraries, or in the case of novel operators, reference implementations are built via DL-framework primitives with underwhelming performance. This work introduces the Tensor Processing Primitives (TPP), a programming abstraction striving for efficient, portable implementation of DL-workloads with high-productivity. TPPs define a compact, yet versatile set of 2D-tensor operators (or a virtual Tensor ISA), which subsequently can be utilized as building-blocks to construct complex operators on high-dimensional tensors. The TPP specification is platform-agnostic, thus code expressed via TPPs is portable, whereas the TPP implementation is highly-optimized and platform-specific. We demonstrate the efficacy of our approach using standalone kernels and end-to-end DL-workloads expressed entirely via TPPs that outperform state-of-the-art implementations on multiple platforms.
Deep Graph Library Optimizations for Intel(R) x86 Architecture
Jul 13, 2020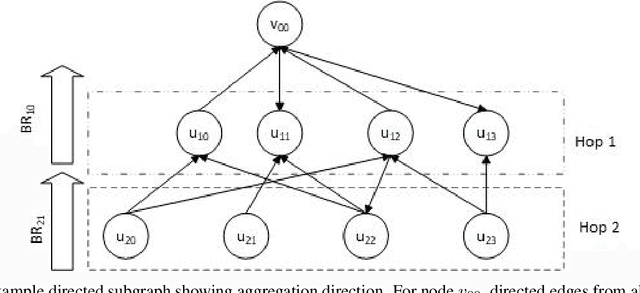
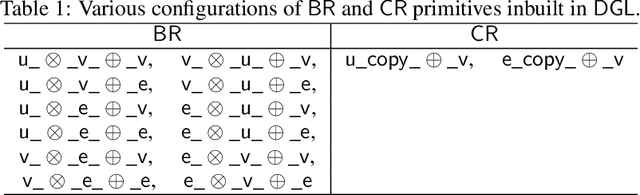
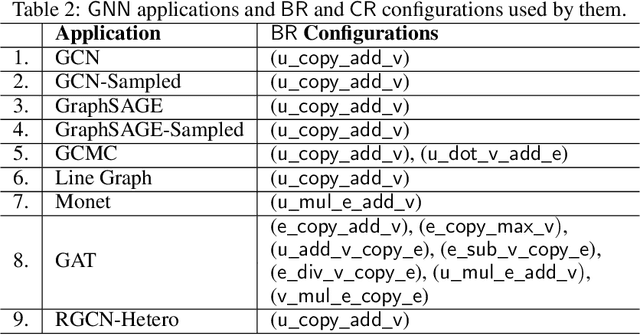
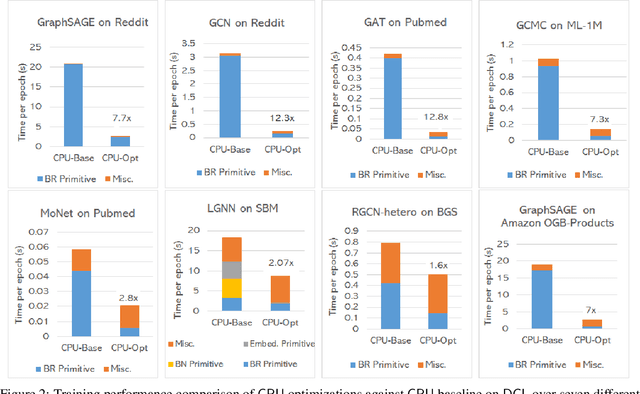
The Deep Graph Library (DGL) was designed as a tool to enable structure learning from graphs, by supporting a core abstraction for graphs, including the popular Graph Neural Networks (GNN). DGL contains implementations of all core graph operations for both the CPU and GPU. In this paper, we focus specifically on CPU implementations and present performance analysis, optimizations and results across a set of GNN applications using the latest version of DGL(0.4.3). Across 7 applications, we achieve speed-ups ranging from1 1.5x-13x over the baseline CPU implementations.
LISA: Towards Learned DNA Sequence Search
Oct 10, 2019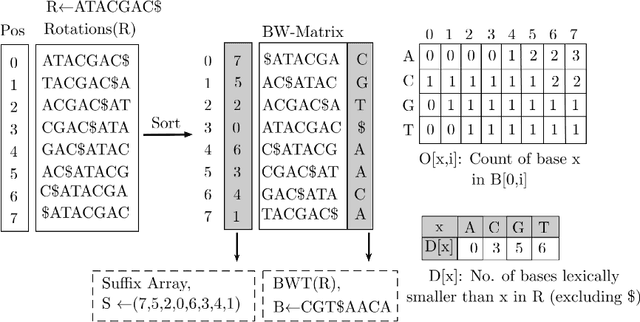

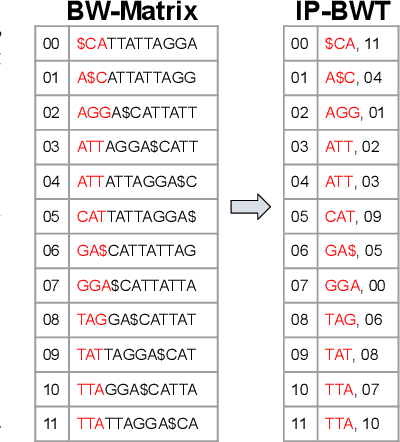
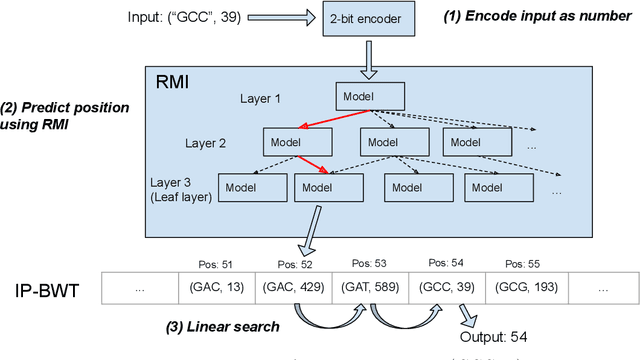
Next-generation sequencing (NGS) technologies have enabled affordable sequencing of billions of short DNA fragments at high throughput, paving the way for population-scale genomics. Genomics data analytics at this scale requires overcoming performance bottlenecks, such as searching for short DNA sequences over long reference sequences. In this paper, we introduce LISA (Learned Indexes for Sequence Analysis), a novel learning-based approach to DNA sequence search. As a first proof of concept, we focus on accelerating one of the most essential flavors of the problem, called exact search. LISA builds on and extends FM-index, which is the state-of-the-art technique widely deployed in genomics tool-chains. Initial experiments with human genome datasets indicate that LISA achieves up to a factor of 4X performance speedup against its traditional counterpart.