 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVeriLLMed: Interactive Visual Debugging of Medical Large Language Models with Knowledge Graphs
Apr 25, 2026Large language models (LLMs) show promise in medical diagnosis, but real-world deployment remains challenging due to high-stakes clinical decisions and imperfect reasoning reliability. As a result, careful inspection of model behavior is essential for assessing whether diagnostic reasoning is reliable and clinically grounded. However, debugging medical LLMs remains difficult. First, developers often lack sufficient medical domain expertise to interpret model errors in clinically meaningful terms. Second, models can fail across a large and diverse set of instances involving different input types, tasks, and reasoning steps, making it challenging for developers to prioritize which errors deserve focused inspection. Third, developers struggle to identify recurring error patterns across cases, as existing debugging practices are largely instance-centric and rely on manual inspection of isolated failures. To address these challenges, we present VeriLLMed, a visual analytics system that integrates external biomedical knowledge to audit and debug medical LLM diagnostic reasoning. VeriLLMed transforms model outputs into comparable reasoning paths, constructs knowledge graph-grounded reference paths, and identifies three recurring classes of diagnosis errors: relation errors, branch errors, and missing errors. Case studies and expert evaluation demonstrate that VeriLLMed helps developers identify clinically implausible reasoning and generate actionable insights that can inform the improvement of medical LLMs.
MAT-Cell: A Multi-Agent Tree-Structured Reasoning Framework for Batch-Level Single-Cell Annotation
Apr 07, 2026Automated cellular reasoning faces a core dichotomy: supervised methods fall into the Reference Trap and fail to generalize to out-of-distribution cell states, while large language models (LLMs), without grounded biological priors, suffer from a Signal-to-Noise Paradox that produces spurious associations. We propose MAT-Cell, a neuro-symbolic reasoning framework that reframes single-cell analysis from black-box classification into constructive, verifiable proof generation. MAT-Cell injects symbolic constraints through adaptive Retrieval-Augmented Generation (RAG) to ground neural reasoning in biological axioms and reduce transcriptomic noise. It further employs a dialectic verification process with homogeneous rebuttal agents to audit and prune reasoning paths, forming syllogistic derivation trees that enforce logical consistency.Across large-scale and cross-species benchmarks, MAT-Cell significantly outperforms state-of-the-art (SOTA) models and maintains robust per-formance in challenging scenarios where baselinemethods severely degrade. Code is available at https://gith ub.com/jiangliu91/MAT-Cell-A-Mul ti-Agent-Tree-Structured-Reasoni ng-Framework-for-Batch-Level-Sin gle-Cell-Annotation.
Revisiting Cross-Attention Mechanisms: Leveraging Beneficial Noise for Domain-Adaptive Learning
Mar 18, 2026Unsupervised Domain Adaptation (UDA) seeks to transfer knowledge from a labeled source domain to an unlabeled target domain but often suffers from severe domain and scale gaps that degrade performance. Existing cross-attention-based transformers can align features across domains, yet they struggle to preserve content semantics under large appearance and scale variations. To explicitly address these challenges, we introduce the concept of beneficial noise, which regularizes cross-attention by injecting controlled perturbations, encouraging the model to ignore style distractions and focus on content. We propose the Domain-Adaptive Cross-Scale Matching (DACSM) framework, which consists of a Domain-Adaptive Transformer (DAT) for disentangling domain-shared content from domain-specific style, and a Cross-Scale Matching (CSM) module that adaptively aligns features across multiple resolutions. DAT incorporates beneficial noise into cross-attention, enabling progressive domain translation with enhanced robustness, yielding content-consistent and style-invariant representations. Meanwhile, CSM ensures semantic consistency under scale changes. Extensive experiments on VisDA-2017, Office-Home, and DomainNet demonstrate that DACSM achieves state-of-the-art performance, with up to +2.3% improvement over CDTrans on VisDA-2017. Notably, DACSM achieves a +5.9% gain on the challenging "truck" class of VisDA, evidencing the strength of beneficial noise in handling scale discrepancies. These results highlight the effectiveness of combining domain translation, beneficial-noise-enhanced attention, and scale-aware alignment for robust cross-domain representation learning.
Latent Poincaré Shaping for Agentic Reinforcement Learning
Feb 10, 2026We propose LaPha, a method for training AlphaZero-like LLM agents in a Poincaré latent space. Under LaPha, the search process can be visualized as a tree rooted at the prompt and growing outward from the origin toward the boundary of the Poincaré ball, where negative curvature provides exponentially increasing capacity with radius. Using hyperbolic geodesic distance to rule-verified correctness, we define a node potential and assign dense process rewards by potential differences. We further attach a lightweight value head on the same shared latent space, enabling self-guided test-time scaling with almost no additional overhead. On MATH-500, LaPha improves Qwen2.5-Math-1.5B from 66.0% to 88.2%. With value-head-guided search, LaPha-1.5B reaches 56.7% accuracy on AIME'24, and LaPha-7B further achieves 60.0% on AIME'24 and 53.3% on AIME'25.
UniSurg: A Video-Native Foundation Model for Universal Understanding of Surgical Videos
Feb 05, 2026While foundation models have advanced surgical video analysis, current approaches rely predominantly on pixel-level reconstruction objectives that waste model capacity on low-level visual details - such as smoke, specular reflections, and fluid motion - rather than semantic structures essential for surgical understanding. We present UniSurg, a video-native foundation model that shifts the learning paradigm from pixel-level reconstruction to latent motion prediction. Built on the Video Joint Embedding Predictive Architecture (V-JEPA), UniSurg introduces three key technical innovations tailored to surgical videos: 1) motion-guided latent prediction to prioritize semantically meaningful regions, 2) spatiotemporal affinity self-distillation to enforce relational consistency, and 3) feature diversity regularization to prevent representation collapse in texture-sparse surgical scenes. To enable large-scale pretraining, we curate UniSurg-15M, the largest surgical video dataset to date, comprising 3,658 hours of video from 50 sources across 13 anatomical regions. Extensive experiments across 17 benchmarks demonstrate that UniSurg significantly outperforms state-of-the-art methods on surgical workflow recognition (+14.6% F1 on EgoSurgery, +10.3% on PitVis), action triplet recognition (39.54% mAP-IVT on CholecT50), skill assessment, polyp segmentation, and depth estimation. These results establish UniSurg as a new standard for universal, motion-oriented surgical video understanding.
A Medical Multimodal Diagnostic Framework Integrating Vision-Language Models and Logic Tree Reasoning
Dec 25, 2025With the rapid growth of large language models (LLMs) and vision-language models (VLMs) in medicine, simply integrating clinical text and medical imaging does not guarantee reliable reasoning. Existing multimodal models often produce hallucinations or inconsistent chains of thought, limiting clinical trust. We propose a diagnostic framework built upon LLaVA that combines vision-language alignment with logic-regularized reasoning. The system includes an input encoder for text and images, a projection module for cross-modal alignment, a reasoning controller that decomposes diagnostic tasks into steps, and a logic tree generator that assembles stepwise premises into verifiable conclusions. Evaluations on MedXpertQA and other benchmarks show that our method improves diagnostic accuracy and yields more interpretable reasoning traces on multimodal tasks, while remaining competitive on text-only settings. These results suggest a promising step toward trustworthy multimodal medical AI.
Anatomy-R1: Enhancing Anatomy Reasoning in Multimodal Large Language Models via Anatomical Similarity Curriculum and Group Diversity Augmentation
Dec 24, 2025Multimodal Large Language Models (MLLMs) have achieved impressive progress in natural image reasoning, yet their potential in medical imaging remains underexplored, especially in clinical anatomical surgical images. Anatomy understanding tasks demand precise understanding and clinically coherent answers, which are difficult to achieve due to the complexity of medical data and the scarcity of high-quality expert annotations. These challenges limit the effectiveness of conventional Supervised Fine-Tuning (SFT) strategies. While recent work has demonstrated that Group Relative Policy Optimization (GRPO) can enhance reasoning in MLLMs without relying on large amounts of data, we find two weaknesses that hinder GRPO's reasoning performance in anatomy recognition: 1) knowledge cannot be effectively shared between different anatomical structures, resulting in uneven information gain and preventing the model from converging, and 2) the model quickly converges to a single reasoning path, suppressing the exploration of diverse strategies. To overcome these challenges, we propose two novel methods. First, we implement a progressive learning strategy called Anatomical Similarity Curriculum Learning by controlling question difficulty via the similarity of answer choices, enabling the model to master complex problems incrementally. Second, we utilize question augmentation referred to as Group Diversity Question Augmentation to expand the model's search space for difficult queries, mitigating the tendency to produce uniform responses. Comprehensive experiments on the SGG-VQA and OmniMedVQA benchmarks show our method achieves a significant improvement across the two benchmarks, demonstrating its effectiveness in enhancing the medical reasoning capabilities of MLLMs. The code can be found in https://github.com/tomato996/Anatomy-R1
Departures: Distributional Transport for Single-Cell Perturbation Prediction with Neural Schrödinger Bridges
Nov 17, 2025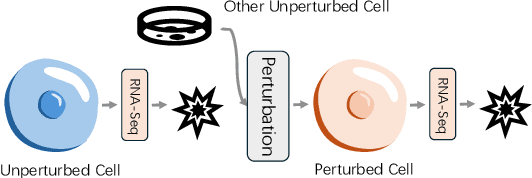
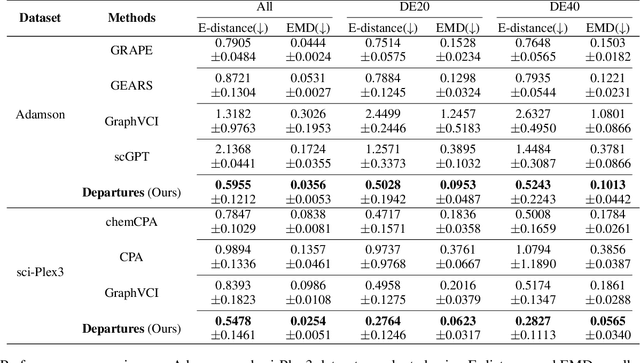
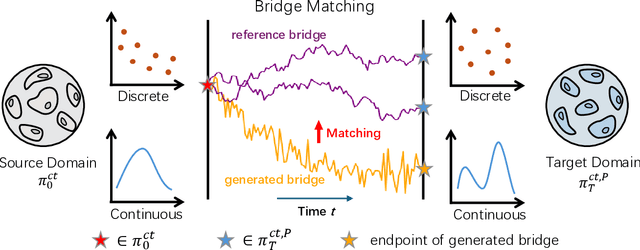

Predicting single-cell perturbation outcomes directly advances gene function analysis and facilitates drug candidate selection, making it a key driver of both basic and translational biomedical research. However, a major bottleneck in this task is the unpaired nature of single-cell data, as the same cell cannot be observed both before and after perturbation due to the destructive nature of sequencing. Although some neural generative transport models attempt to tackle unpaired single-cell perturbation data, they either lack explicit conditioning or depend on prior spaces for indirect distribution alignment, limiting precise perturbation modeling. In this work, we approximate Schrödinger Bridge (SB), which defines stochastic dynamic mappings recovering the entropy-regularized optimal transport (OT), to directly align the distributions of control and perturbed single-cell populations across different perturbation conditions. Unlike prior SB approximations that rely on bidirectional modeling to infer optimal source-target sample coupling, we leverage Minibatch-OT based pairing to avoid such bidirectional inference and the associated ill-posedness of defining the reverse process. This pairing directly guides bridge learning, yielding a scalable approximation to the SB. We approximate two SB models, one modeling discrete gene activation states and the other continuous expression distributions. Joint training enables accurate perturbation modeling and captures single-cell heterogeneity. Experiments on public genetic and drug perturbation datasets show that our model effectively captures heterogeneous single-cell responses and achieves state-of-the-art performance.
PhyloGen: Language Model-Enhanced Phylogenetic Inference via Graph Structure Generation
Dec 25, 2024



Phylogenetic trees elucidate evolutionary relationships among species, but phylogenetic inference remains challenging due to the complexity of combining continuous (branch lengths) and discrete parameters (tree topology). Traditional Markov Chain Monte Carlo methods face slow convergence and computational burdens. Existing Variational Inference methods, which require pre-generated topologies and typically treat tree structures and branch lengths independently, may overlook critical sequence features, limiting their accuracy and flexibility. We propose PhyloGen, a novel method leveraging a pre-trained genomic language model to generate and optimize phylogenetic trees without dependence on evolutionary models or aligned sequence constraints. PhyloGen views phylogenetic inference as a conditionally constrained tree structure generation problem, jointly optimizing tree topology and branch lengths through three core modules: (i) Feature Extraction, (ii) PhyloTree Construction, and (iii) PhyloTree Structure Modeling. Meanwhile, we introduce a Scoring Function to guide the model towards a more stable gradient descent. We demonstrate the effectiveness and robustness of PhyloGen on eight real-world benchmark datasets. Visualization results confirm PhyloGen provides deeper insights into phylogenetic relationships.
DMT-HI: MOE-based Hyperbolic Interpretable Deep Manifold Transformation for Unspervised Dimensionality Reduction
Oct 25, 2024



Dimensionality reduction (DR) plays a crucial role in various fields, including data engineering and visualization, by simplifying complex datasets while retaining essential information. However, the challenge of balancing DR accuracy and interpretability remains crucial, particularly for users dealing with high-dimensional data. Traditional DR methods often face a trade-off between precision and transparency, where optimizing for performance can lead to reduced interpretability, and vice versa. This limitation is especially prominent in real-world applications such as image, tabular, and text data analysis, where both accuracy and interpretability are critical. To address these challenges, this work introduces the MOE-based Hyperbolic Interpretable Deep Manifold Transformation (DMT-HI). The proposed approach combines hyperbolic embeddings, which effectively capture complex hierarchical structures, with Mixture of Experts (MOE) models, which dynamically allocate tasks based on input features. DMT-HI enhances DR accuracy by leveraging hyperbolic embeddings to represent the hierarchical nature of data, while also improving interpretability by explicitly linking input data, embedding outcomes, and key features through the MOE structure. Extensive experiments demonstrate that DMT-HI consistently achieves superior performance in both DR accuracy and model interpretability, making it a robust solution for complex data analysis. The code is available at \url{https://github.com/zangzelin/code_dmthi}.