 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDiffVC: A Non-autoregressive Framework Based on Diffusion Model for Video Captioning
Apr 09, 2026Current video captioning methods usually use an encoder-decoder structure to generate text autoregressively. However, autoregressive methods have inherent limitations such as slow generation speed and large cumulative error. Furthermore, the few non-autoregressive counterparts suffer from deficiencies in generation quality due to the lack of sufficient multimodal interaction modeling. Therefore, we propose a non-autoregressive framework based on Diffusion model for Video Captioning (DiffVC) to address these issues. Its parallel decoding can effectively solve the problems of generation speed and cumulative error. At the same time, our proposed discriminative conditional Diffusion Model can generate higher-quality textual descriptions. Specifically, we first encode the video into a visual representation. During training, Gaussian noise is added to the textual representation of the ground-truth caption. Then, a new textual representation is generated via the discriminative denoiser with the visual representation as a conditional constraint. Finally, we input the new textual representation into a non-autoregressive language model to generate captions. During inference, we directly sample noise from the Gaussian distribution for generation. Experiments on MSVD, MSR-VTT, and VATEX show that our method can outperform previous non-autoregressive methods and achieve comparable performance to autoregressive methods, e.g., it achieved a maximum improvement of 9.9 on the CIDEr and improvement of 2.6 on the B@4, while having faster generation speed. The source code will be available soon.
Probing Scientific General Intelligence of LLMs with Scientist-Aligned Workflows
Dec 18, 2025Despite advances in scientific AI, a coherent framework for Scientific General Intelligence (SGI)-the ability to autonomously conceive, investigate, and reason across scientific domains-remains lacking. We present an operational SGI definition grounded in the Practical Inquiry Model (PIM: Deliberation, Conception, Action, Perception) and operationalize it via four scientist-aligned tasks: deep research, idea generation, dry/wet experiments, and experimental reasoning. SGI-Bench comprises over 1,000 expert-curated, cross-disciplinary samples inspired by Science's 125 Big Questions, enabling systematic evaluation of state-of-the-art LLMs. Results reveal gaps: low exact match (10--20%) in deep research despite step-level alignment; ideas lacking feasibility and detail; high code executability but low execution result accuracy in dry experiments; low sequence fidelity in wet protocols; and persistent multimodal comparative-reasoning challenges. We further introduce Test-Time Reinforcement Learning (TTRL), which optimizes retrieval-augmented novelty rewards at inference, enhancing hypothesis novelty without reference answer. Together, our PIM-grounded definition, workflow-centric benchmark, and empirical insights establish a foundation for AI systems that genuinely participate in scientific discovery.
Departures: Distributional Transport for Single-Cell Perturbation Prediction with Neural Schrödinger Bridges
Nov 17, 2025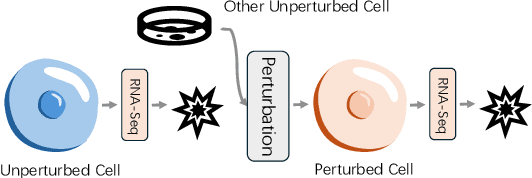
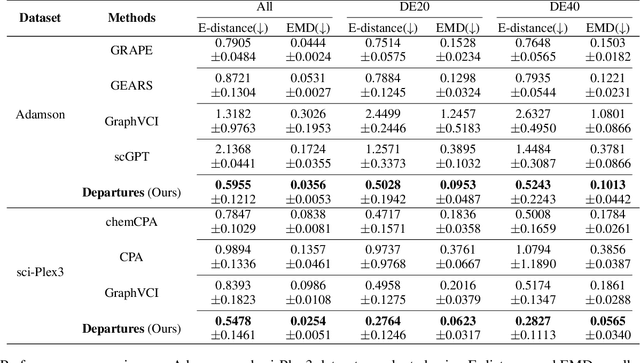
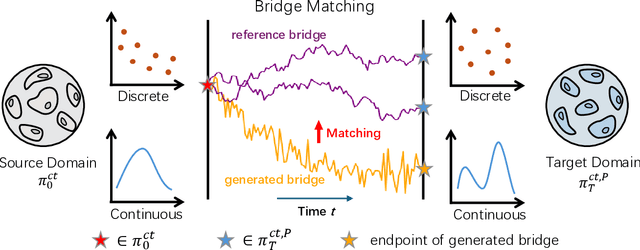

Predicting single-cell perturbation outcomes directly advances gene function analysis and facilitates drug candidate selection, making it a key driver of both basic and translational biomedical research. However, a major bottleneck in this task is the unpaired nature of single-cell data, as the same cell cannot be observed both before and after perturbation due to the destructive nature of sequencing. Although some neural generative transport models attempt to tackle unpaired single-cell perturbation data, they either lack explicit conditioning or depend on prior spaces for indirect distribution alignment, limiting precise perturbation modeling. In this work, we approximate Schrödinger Bridge (SB), which defines stochastic dynamic mappings recovering the entropy-regularized optimal transport (OT), to directly align the distributions of control and perturbed single-cell populations across different perturbation conditions. Unlike prior SB approximations that rely on bidirectional modeling to infer optimal source-target sample coupling, we leverage Minibatch-OT based pairing to avoid such bidirectional inference and the associated ill-posedness of defining the reverse process. This pairing directly guides bridge learning, yielding a scalable approximation to the SB. We approximate two SB models, one modeling discrete gene activation states and the other continuous expression distributions. Joint training enables accurate perturbation modeling and captures single-cell heterogeneity. Experiments on public genetic and drug perturbation datasets show that our model effectively captures heterogeneous single-cell responses and achieves state-of-the-art performance.
10K is Enough: An Ultra-Lightweight Binarized Network for Infrared Small-Target Detection
Mar 04, 2025
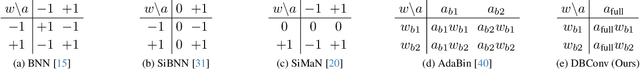
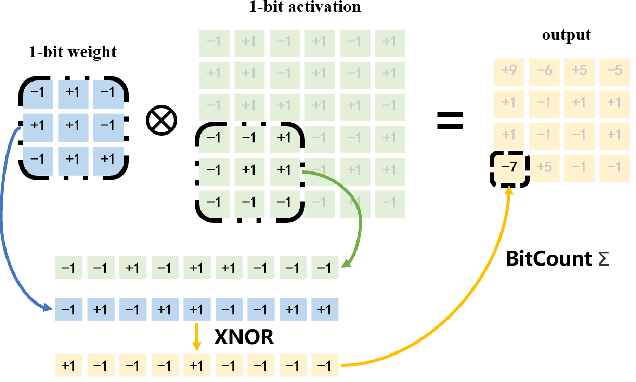
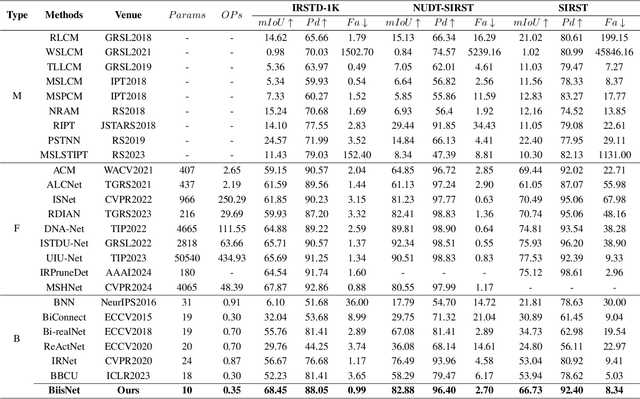
The widespread deployment of InfRared Small-Target Detection(IRSTD) algorithms on edge devices necessitates the exploration of model compression techniques. Binary neural networks (BNNs) are distinguished by their exceptional efficiency in model compression. However, the small size of infrared targets introduces stringent precision requirements for the IRSTD task, while the inherent precision loss during binarization presents a significant challenge. To address this, we propose the Binarized Infrared Small-Target Detection Network (BiisNet), which preserves the core operations of binarized convolutions while integrating full-precision features into the network's information flow. Specifically, we propose the Dot-Binary Convolution, which retains fine-grained semantic information in feature maps while still leveraging the binarized convolution operations. In addition, we introduce a smooth and adaptive Dynamic Softsign function, which provides more comprehensive and progressively finer gradient during back-propagation, enhancing model stability and promoting an optimal weight distribution.Experimental results demonstrate that BiisNet not only significantly outperforms other binary architectures but also demonstrates strong competitiveness among state-of-the-art full-precision models.
DapPep: Domain Adaptive Peptide-agnostic Learning for Universal T-cell Receptor-antigen Binding Affinity Prediction
Nov 26, 2024


Identifying T-cell receptors (TCRs) that interact with antigenic peptides provides the technical basis for developing vaccines and immunotherapies. The emergent deep learning methods excel at learning antigen binding patterns from known TCRs but struggle with novel or sparsely represented antigens. However, binding specificity for unseen antigens or exogenous peptides is critical. We introduce a domain-adaptive peptide-agnostic learning framework DapPep for universal TCR-antigen binding affinity prediction to address this challenge. The lightweight self-attention architecture combines a pre-trained protein language model with an inner-loop self-supervised regime to enable robust TCR-peptide representations. Extensive experiments on various benchmarks demonstrate that DapPep consistently outperforms existing tools, showcasing robust generalization capability, especially for data-scarce settings and unseen peptides. Moreover, DapPep proves effective in challenging clinical tasks such as sorting reactive T cells in tumor neoantigen therapy and identifying key positions in 3D structures.
Pan-protein Design Learning Enables Task-adaptive Generalization for Low-resource Enzyme Design
Nov 26, 2024Computational protein design (CPD) offers transformative potential for bioengineering, but current deep CPD models, focused on universal domains, struggle with function-specific designs. This work introduces a novel CPD paradigm tailored for functional design tasks, particularly for enzymes-a key protein class often lacking specific application efficiency. To address structural data scarcity, we present CrossDesign, a domain-adaptive framework that leverages pretrained protein language models (PPLMs). By aligning protein structures with sequences, CrossDesign transfers pretrained knowledge to structure models, overcoming the limitations of limited structural data. The framework combines autoregressive (AR) and non-autoregressive (NAR) states in its encoder-decoder architecture, applying it to enzyme datasets and pan-proteins. Experimental results highlight CrossDesign's superior performance and robustness, especially with out-of-domain enzymes. Additionally, the model excels in fitness prediction when tested on large-scale mutation data, showcasing its stability.
MetaEnzyme: Meta Pan-Enzyme Learning for Task-Adaptive Redesign
Aug 05, 2024



Enzyme design plays a crucial role in both industrial production and biology. However, this field faces challenges due to the lack of comprehensive benchmarks and the complexity of enzyme design tasks, leading to a dearth of systematic research. Consequently, computational enzyme design is relatively overlooked within the broader protein domain and remains in its early stages. In this work, we address these challenges by introducing MetaEnzyme, a staged and unified enzyme design framework. We begin by employing a cross-modal structure-to-sequence transformation architecture, as the feature-driven starting point to obtain initial robust protein representation. Subsequently, we leverage domain adaptive techniques to generalize specific enzyme design tasks under low-resource conditions. MetaEnzyme focuses on three fundamental low-resource enzyme redesign tasks: functional design (FuncDesign), mutation design (MutDesign), and sequence generation design (SeqDesign). Through novel unified paradigm and enhanced representation capabilities, MetaEnzyme demonstrates adaptability to diverse enzyme design tasks, yielding outstanding results. Wet lab experiments further validate these findings, reinforcing the efficacy of the redesign process.
Gentle-CLIP: Exploring Aligned Semantic In Low-Quality Multimodal Data With Soft Alignment
Jun 09, 2024Multimodal fusion breaks through the barriers between diverse modalities and has already yielded numerous impressive performances. However, in various specialized fields, it is struggling to obtain sufficient alignment data for the training process, which seriously limits the use of previously elegant models. Thus, semi-supervised learning attempts to achieve multimodal alignment with fewer matched pairs but traditional methods like pseudo-labeling are difficult to apply in domains with no label information. To address these problems, we transform semi-supervised multimodal alignment into a manifold matching problem and propose a new method based on CLIP, named Gentle-CLIP. Specifically, we design a novel semantic density distribution loss to explore implicit semantic alignment information from unpaired multimodal data by constraining the latent representation distribution with fine granularity, thus eliminating the need for numerous strictly matched pairs. Meanwhile, we introduce multi-kernel maximum mean discrepancy as well as self-supervised contrastive loss to pull separate modality distributions closer and enhance the stability of the representation distribution. In addition, the contrastive loss used in CLIP is employed on the supervised matched data to prevent negative optimization. Extensive experiments conducted on a range of tasks in various fields, including protein, remote sensing, and the general vision-language field, demonstrate the effectiveness of our proposed Gentle-CLIP.
VQDNA: Unleashing the Power of Vector Quantization for Multi-Species Genomic Sequence Modeling
May 13, 2024Similar to natural language models, pre-trained genome language models are proposed to capture the underlying intricacies within genomes with unsupervised sequence modeling. They have become essential tools for researchers and practitioners in biology. However, the \textit{hand-crafted} tokenization policies used in these models may not encode the most discriminative patterns from the limited vocabulary of genomic data. In this paper, we introduce VQDNA, a general-purpose framework that renovates genome tokenization from the perspective of genome vocabulary learning. By leveraging vector-quantized codebook as \textit{learnable} vocabulary, VQDNA can adaptively tokenize genomes into \textit{pattern-aware} embeddings in an end-to-end manner. To further push its limits, we propose Hierarchical Residual Quantization (HRQ), where varying scales of codebooks are designed in a hierarchy to enrich the genome vocabulary in a coarse-to-fine manner. Extensive experiments on 32 genome datasets demonstrate VQDNA's superiority and favorable parameter efficiency compared to existing genome language models. Notably, empirical analysis of SARS-CoV-2 mutations reveals the fine-grained pattern awareness and biological significance of learned HRQ vocabulary, highlighting its untapped potential for broader applications in genomics.
Inter- and intra-uncertainty based feature aggregation model for semi-supervised histopathology image segmentation
Mar 19, 2024Acquiring pixel-level annotations is often limited in applications such as histology studies that require domain expertise. Various semi-supervised learning approaches have been developed to work with limited ground truth annotations, such as the popular teacher-student models. However, hierarchical prediction uncertainty within the student model (intra-uncertainty) and image prediction uncertainty (inter-uncertainty) have not been fully utilized by existing methods. To address these issues, we first propose a novel inter- and intra-uncertainty regularization method to measure and constrain both inter- and intra-inconsistencies in the teacher-student architecture. We also propose a new two-stage network with pseudo-mask guided feature aggregation (PG-FANet) as the segmentation model. The two-stage structure complements with the uncertainty regularization strategy to avoid introducing extra modules in solving uncertainties and the aggregation mechanisms enable multi-scale and multi-stage feature integration. Comprehensive experimental results over the MoNuSeg and CRAG datasets show that our PG-FANet outperforms other state-of-the-art methods and our semi-supervised learning framework yields competitive performance with a limited amount of labeled data.