 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhyloGen: Language Model-Enhanced Phylogenetic Inference via Graph Structure Generation
Dec 25, 2024



Phylogenetic trees elucidate evolutionary relationships among species, but phylogenetic inference remains challenging due to the complexity of combining continuous (branch lengths) and discrete parameters (tree topology). Traditional Markov Chain Monte Carlo methods face slow convergence and computational burdens. Existing Variational Inference methods, which require pre-generated topologies and typically treat tree structures and branch lengths independently, may overlook critical sequence features, limiting their accuracy and flexibility. We propose PhyloGen, a novel method leveraging a pre-trained genomic language model to generate and optimize phylogenetic trees without dependence on evolutionary models or aligned sequence constraints. PhyloGen views phylogenetic inference as a conditionally constrained tree structure generation problem, jointly optimizing tree topology and branch lengths through three core modules: (i) Feature Extraction, (ii) PhyloTree Construction, and (iii) PhyloTree Structure Modeling. Meanwhile, we introduce a Scoring Function to guide the model towards a more stable gradient descent. We demonstrate the effectiveness and robustness of PhyloGen on eight real-world benchmark datasets. Visualization results confirm PhyloGen provides deeper insights into phylogenetic relationships.
LaserGuider: A Laser Based Physical Backdoor Attack against Deep Neural Networks
Dec 05, 2024



Backdoor attacks embed hidden associations between triggers and targets in deep neural networks (DNNs), causing them to predict the target when a trigger is present while maintaining normal behavior otherwise. Physical backdoor attacks, which use physical objects as triggers, are feasible but lack remote control, temporal stealthiness, flexibility, and mobility. To overcome these limitations, in this work, we propose a new type of backdoor triggers utilizing lasers that feature long-distance transmission and instant-imaging properties. Based on the laser-based backdoor triggers, we present a physical backdoor attack, called LaserGuider, which possesses remote control ability and achieves high temporal stealthiness, flexibility, and mobility. We also introduce a systematic approach to optimize laser parameters for improving attack effectiveness. Our evaluation on traffic sign recognition DNNs, critical in autonomous vehicles, demonstrates that LaserGuider with three different laser-based triggers achieves over 90% attack success rate with negligible impact on normal inputs. Additionally, we release LaserMark, the first dataset of real world traffic signs stamped with physical laser spots, to support further research in backdoor attacks and defenses.
A Review of Artificial Intelligence based Biological-Tree Construction: Priorities, Methods, Applications and Trends
Oct 07, 2024
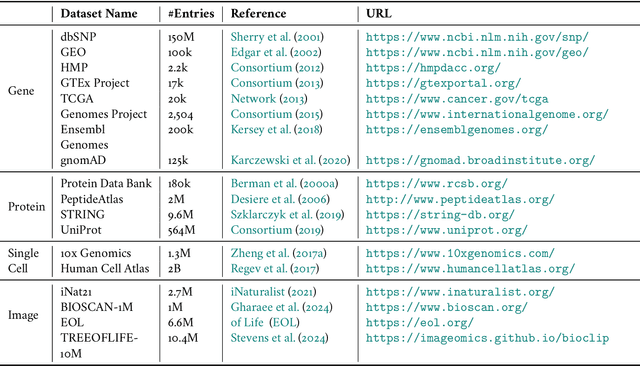
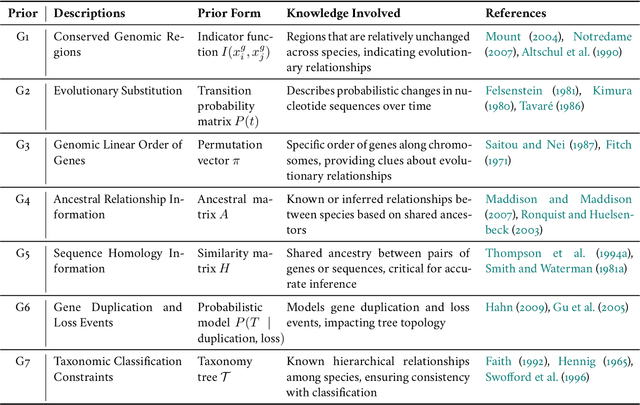

Biological tree analysis serves as a pivotal tool in uncovering the evolutionary and differentiation relationships among organisms, genes, and cells. Its applications span diverse fields including phylogenetics, developmental biology, ecology, and medicine. Traditional tree inference methods, while foundational in early studies, face increasing limitations in processing the large-scale, complex datasets generated by modern high-throughput technologies. Recent advances in deep learning offer promising solutions, providing enhanced data processing and pattern recognition capabilities. However, challenges remain, particularly in accurately representing the inherently discrete and non-Euclidean nature of biological trees. In this review, we first outline the key biological priors fundamental to phylogenetic and differentiation tree analyses, facilitating a deeper interdisciplinary understanding between deep learning researchers and biologists. We then systematically examine the commonly used data formats and databases, serving as a comprehensive resource for model testing and development. We provide a critical analysis of traditional tree generation methods, exploring their underlying biological assumptions, technical characteristics, and limitations. Current developments in deep learning-based tree generation are reviewed, highlighting both recent advancements and existing challenges. Furthermore, we discuss the diverse applications of biological trees across various biological domains. Finally, we propose potential future directions and trends in leveraging deep learning for biological tree research, aiming to guide further exploration and innovation in this field.
FGBERT: Function-Driven Pre-trained Gene Language Model for Metagenomics
Feb 24, 2024



Metagenomic data, comprising mixed multi-species genomes, are prevalent in diverse environments like oceans and soils, significantly impacting human health and ecological functions. However, current research relies on K-mer representations, limiting the capture of structurally relevant gene contexts. To address these limitations and further our understanding of complex relationships between metagenomic sequences and their functions, we introduce a protein-based gene representation as a context-aware and structure-relevant tokenizer. Our approach includes Masked Gene Modeling (MGM) for gene group-level pre-training, providing insights into inter-gene contextual information, and Triple Enhanced Metagenomic Contrastive Learning (TEM-CL) for gene-level pre-training to model gene sequence-function relationships. MGM and TEM-CL constitute our novel metagenomic language model {\NAME}, pre-trained on 100 million metagenomic sequences. We demonstrate the superiority of our proposed {\NAME} on eight datasets.
Must: Maximizing Latent Capacity of Spatial Transcriptomics Data
Jan 15, 2024Spatial transcriptomics (ST) technologies have revolutionized the study of gene expression patterns in tissues by providing multimodality data in transcriptomic, spatial, and morphological, offering opportunities for understanding tissue biology beyond transcriptomics. However, we identify the modality bias phenomenon in ST data species, i.e., the inconsistent contribution of different modalities to the labels leads to a tendency for the analysis methods to retain the information of the dominant modality. How to mitigate the adverse effects of modality bias to satisfy various downstream tasks remains a fundamental challenge. This paper introduces Multiple-modality Structure Transformation, named MuST, a novel methodology to tackle the challenge. MuST integrates the multi-modality information contained in the ST data effectively into a uniform latent space to provide a foundation for all the downstream tasks. It learns intrinsic local structures by topology discovery strategy and topology fusion loss function to solve the inconsistencies among different modalities. Thus, these topology-based and deep learning techniques provide a solid foundation for a variety of analytical tasks while coordinating different modalities. The effectiveness of MuST is assessed by performance metrics and biological significance. The results show that it outperforms existing state-of-the-art methods with clear advantages in the precision of identifying and preserving structures of tissues and biomarkers. MuST offers a versatile toolkit for the intricate analysis of complex biological systems.
Non-equispaced Fourier Neural Solvers for PDEs
Dec 09, 2022Solving partial differential equations is difficult. Recently proposed neural resolution-invariant models, despite their effectiveness and efficiency, usually require equispaced spatial points of data. However, sampling in spatial domain is sometimes inevitably non-equispaced in real-world systems, limiting their applicability. In this paper, we propose a Non-equispaced Fourier PDE Solver (\textsc{NFS}) with adaptive interpolation on resampled equispaced points and a variant of Fourier Neural Operators as its components. Experimental results on complex PDEs demonstrate its advantages in accuracy and efficiency. Compared with the spatially-equispaced benchmark methods, it achieves superior performance with $42.85\%$ improvements on MAE, and is able to handle non-equispaced data with a tiny loss of accuracy. Besides, to our best knowledge, \textsc{NFS} is the first ML-based method with mesh invariant inference ability to successfully model turbulent flows in non-equispaced scenarios, with a minor deviation of the error on unseen spatial points.
Protein Language Models and Structure Prediction: Connection and Progression
Nov 30, 2022



The prediction of protein structures from sequences is an important task for function prediction, drug design, and related biological processes understanding. Recent advances have proved the power of language models (LMs) in processing the protein sequence databases, which inherit the advantages of attention networks and capture useful information in learning representations for proteins. The past two years have witnessed remarkable success in tertiary protein structure prediction (PSP), including evolution-based and single-sequence-based PSP. It seems that instead of using energy-based models and sampling procedures, protein language model (pLM)-based pipelines have emerged as mainstream paradigms in PSP. Despite the fruitful progress, the PSP community needs a systematic and up-to-date survey to help bridge the gap between LMs in the natural language processing (NLP) and PSP domains and introduce their methodologies, advancements and practical applications. To this end, in this paper, we first introduce the similarities between protein and human languages that allow LMs extended to pLMs, and applied to protein databases. Then, we systematically review recent advances in LMs and pLMs from the perspectives of network architectures, pre-training strategies, applications, and commonly-used protein databases. Next, different types of methods for PSP are discussed, particularly how the pLM-based architectures function in the process of protein folding. Finally, we identify challenges faced by the PSP community and foresee promising research directions along with the advances of pLMs. This survey aims to be a hands-on guide for researchers to understand PSP methods, develop pLMs and tackle challenging problems in this field for practical purposes.
EVNet: An Explainable Deep Network for Dimension Reduction
Nov 21, 2022



Dimension reduction (DR) is commonly utilized to capture the intrinsic structure and transform high-dimensional data into low-dimensional space while retaining meaningful properties of the original data. It is used in various applications, such as image recognition, single-cell sequencing analysis, and biomarker discovery. However, contemporary parametric-free and parametric DR techniques suffer from several significant shortcomings, such as the inability to preserve global and local features and the pool generalization performance. On the other hand, regarding explainability, it is crucial to comprehend the embedding process, especially the contribution of each part to the embedding process, while understanding how each feature affects the embedding results that identify critical components and help diagnose the embedding process. To address these problems, we have developed a deep neural network method called EVNet, which provides not only excellent performance in structural maintainability but also explainability to the DR therein. EVNet starts with data augmentation and a manifold-based loss function to improve embedding performance. The explanation is based on saliency maps and aims to examine the trained EVNet parameters and contributions of components during the embedding process. The proposed techniques are integrated with a visual interface to help the user to adjust EVNet to achieve better DR performance and explainability. The interactive visual interface makes it easier to illustrate the data features, compare different DR techniques, and investigate DR. An in-depth experimental comparison shows that EVNet consistently outperforms the state-of-the-art methods in both performance measures and explainability.
UDRN: Unified Dimensional Reduction Neural Network for Feature Selection and Feature Projection
Jul 08, 2022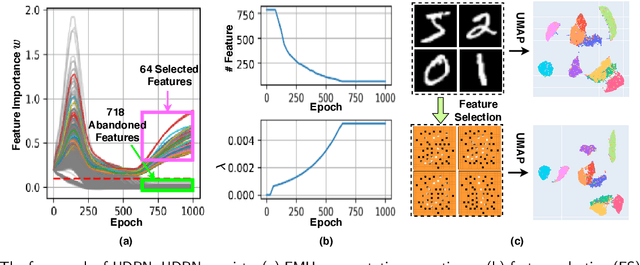
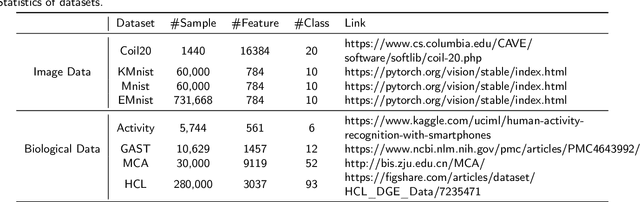
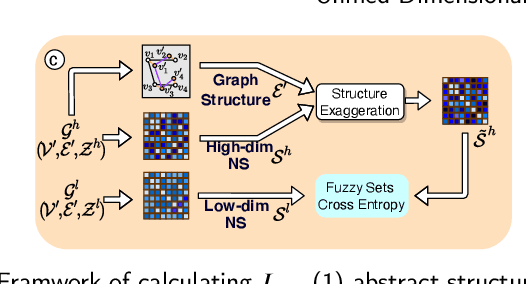

Dimensional reduction~(DR) maps high-dimensional data into a lower dimensions latent space with minimized defined optimization objectives. The DR method usually falls into feature selection~(FS) and feature projection~(FP). FS focuses on selecting a critical subset of dimensions but risks destroying the data distribution (structure). On the other hand, FP combines all the input features into lower dimensions space, aiming to maintain the data structure; but lacks interpretability and sparsity. FS and FP are traditionally incompatible categories; thus, they have not been unified into an amicable framework. We propose that the ideal DR approach combines both FS and FP into a unified end-to-end manifold learning framework, simultaneously performing fundamental feature discovery while maintaining the intrinsic relationships between data samples in the latent space. In this work, we develop a unified framework, Unified Dimensional Reduction Neural-network~(UDRN), that integrates FS and FP in a compatible, end-to-end way. We improve the neural network structure by implementing FS and FP tasks separately using two stacked sub-networks. In addition, we designed data augmentation of the DR process to improve the generalization ability of the method when dealing with extensive feature datasets and designed loss functions that can cooperate with the data augmentation. Extensive experimental results on four image and four biological datasets, including very high-dimensional data, demonstrate the advantages of DRN over existing methods~(FS, FP, and FS\&FP pipeline), especially in downstream tasks such as classification and visualization.
Temporal Attention Unit: Towards Efficient Spatiotemporal Predictive Learning
Jun 24, 2022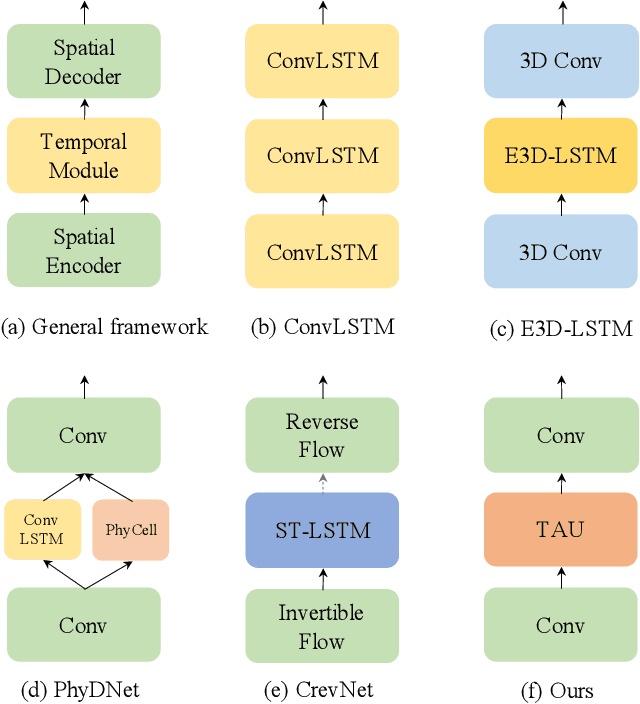
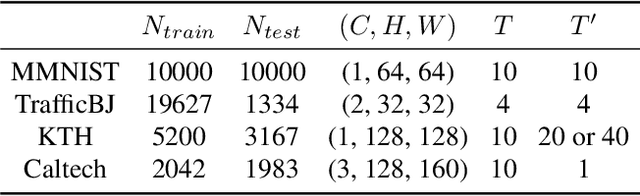
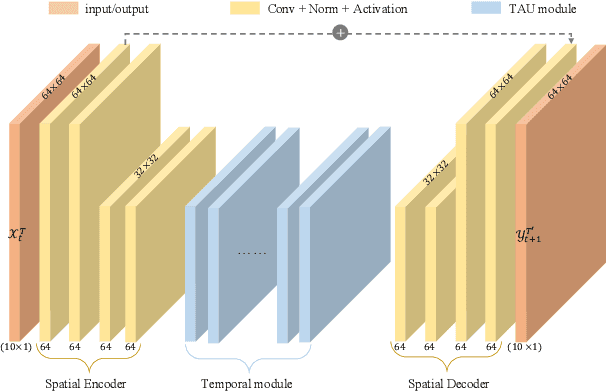
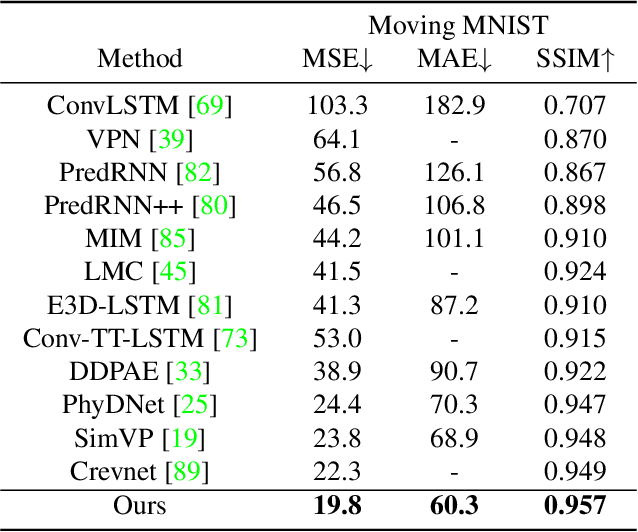
Spatiotemporal predictive learning aims to generate future frames by learning from historical frames. In this paper, we investigate existing methods and present a general framework of spatiotemporal predictive learning, in which the spatial encoder and decoder capture intra-frame features and the middle temporal module catches inter-frame correlations. While the mainstream methods employ recurrent units to capture long-term temporal dependencies, they suffer from low computational efficiency due to their unparallelizable architectures. To parallelize the temporal module, we propose the Temporal Attention Unit (TAU), which decomposes the temporal attention into intra-frame statical attention and inter-frame dynamical attention. Moreover, while the mean squared error loss focuses on intra-frame errors, we introduce a novel differential divergence regularization to take inter-frame variations into account. Extensive experiments demonstrate that the proposed method enables the derived model to achieve competitive performance on various spatiotemporal prediction benchmarks.