 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCan a Lightweight Automated AI Pipeline Solve Research-Level Mathematical Problems?
Feb 14, 2026Large language models (LLMs) have recently achieved remarkable success in generating rigorous mathematical proofs, with "AI for Math" emerging as a vibrant field of research. While these models have mastered competition-level benchmarks like the International Mathematical Olympiad and show promise in research applications through auto-formalization, their deployment via lightweight, natural-language pipelines for research problems remains underexplored. In this work, we demonstrate that next-generation models (e.g., Gemini 3 Pro, GPT-5.2 Pro), when integrated into a streamlined automated pipeline optimized for citation-based verification, can solve sophisticated research-grade problems. We evaluate our pipeline on two novel datasets: (1) the ICCM problem sets (comparable to the S.-T. Yau College Student Mathematics Contest) proposed by leading mathematicians, and (2) the "First Proof" problem set, consisting of previously unpublished research questions. Our pipeline generated candidate proofs for all problems in the first two ICCM sets and the "First Proof" set. The solutions for the first two ICCM sets and Problem 4 of the "First Proof" set have been fully verified by our team. All generated proofs have been submitted to the official organization, and our generated results are publicly available. We plan to open-source the complete pipeline methodology in due course.
GUIGuard: Toward a General Framework for Privacy-Preserving GUI Agents
Jan 26, 2026GUI agents enable end-to-end automation through direct perception of and interaction with on-screen interfaces. However, these agents frequently access interfaces containing sensitive personal information, and screenshots are often transmitted to remote models, creating substantial privacy risks. These risks are particularly severe in GUI workflows: GUIs expose richer, more accessible private information, and privacy risks depend on interaction trajectories across sequential scenes. We propose GUIGuard, a three-stage framework for privacy-preserving GUI agents: (1) privacy recognition, (2) privacy protection, and (3) task execution under protection. We further construct GUIGuard-Bench, a cross-platform benchmark with 630 trajectories and 13,830 screenshots, annotated with region-level privacy grounding and fine-grained labels of risk level, privacy category, and task necessity. Evaluations reveal that existing agents exhibit limited privacy recognition, with state-of-the-art models achieving only 13.3% accuracy on Android and 1.4% on PC. Under privacy protection, task-planning semantics can still be maintained, with closed-source models showing stronger semantic consistency than open-source ones. Case studies on MobileWorld show that carefully designed protection strategies achieve higher task accuracy while preserving privacy. Our results highlight privacy recognition as a critical bottleneck for practical GUI agents. Project: https://futuresis.github.io/GUIGuard-page/
One Tool Is Enough: Reinforcement Learning for Repository-Level LLM Agents
Dec 24, 2025Locating the files and functions requiring modification in large open-source software (OSS) repositories is challenging due to their scale and structural complexity. Existing large language model (LLM)-based methods typically treat this as a repository-level retrieval task and rely on multiple auxiliary tools, which overlook code execution logic and complicate model control. We propose RepoNavigator, an LLM agent equipped with a single execution-aware tool-jumping to the definition of an invoked symbol. This unified design reflects the actual flow of code execution while simplifying tool manipulation. RepoNavigator is trained end-to-end via Reinforcement Learning (RL) directly from a pretrained model, without any closed-source distillation. Experiments demonstrate that RL-trained RepoNavigator achieves state-of-the-art performance, with the 7B model outperforming 14B baselines, the 14B model surpassing 32B competitors, and even the 32B model exceeding closed-source models such as Claude-3.7. These results confirm that integrating a single, structurally grounded tool with RL training provides an efficient and scalable solution for repository-level issue localization.
Population-Evolve: a Parallel Sampling and Evolutionary Method for LLM Math Reasoning
Dec 22, 2025Test-time scaling has emerged as a promising direction for enhancing the reasoning capabilities of Large Language Models in last few years. In this work, we propose Population-Evolve, a training-free method inspired by Genetic Algorithms to optimize LLM reasoning. Our approach maintains a dynamic population of candidate solutions for each problem via parallel reasoning. By incorporating an evolve prompt, the LLM self-evolves its population in all iterations. Upon convergence, the final answer is derived via majority voting. Furthermore, we establish a unification framework that interprets existing test-time scaling strategies through the lens of genetic algorithms. Empirical results demonstrate that Population-Evolve achieves superior accuracy with low performance variance and computational efficiency. Our findings highlight the potential of evolutionary strategies to unlock the reasoning power of LLMs during inference.
RedChronos: A Large Language Model-Based Log Analysis System for Insider Threat Detection in Enterprises
Mar 04, 2025Internal threat detection aims to address security threats within organizations or enterprises by identifying potential or already occurring malicious threats within vast amounts of logs. Although organizations or enterprises have dedicated personnel responsible for reviewing these logs, it is impossible to manually examine all logs entirely. In response to the vast number of logs, we propose a system called RedChronos, which is a Large Language Model-Based Log Analysis System. This system incorporates innovative improvements over previous research by employing Query-Aware Weighted Voting and a Semantic Expansion-based Genetic Algorithm with LLM-driven Mutations. On the public datasets CERT 4.2 and 5.2, RedChronos outperforms or matches existing approaches in terms of accuracy, precision, and detection rate. Moreover, RedChronos reduces the need for manual intervention in security log reviews by 90\% in the Xiaohongshu SOC. Therefore, our RedChronos system demonstrates exceptional performance in handling Internal Threat Detection (IDT) tasks, providing innovative solutions for these challenges. We believe that future research can continue to enhance the system's performance in IDT tasks while also reducing the response time to internal risk events.
Physical Consistency Bridges Heterogeneous Data in Molecular Multi-Task Learning
Oct 14, 2024



In recent years, machine learning has demonstrated impressive capability in handling molecular science tasks. To support various molecular properties at scale, machine learning models are trained in the multi-task learning paradigm. Nevertheless, data of different molecular properties are often not aligned: some quantities, e.g. equilibrium structure, demand more cost to compute than others, e.g. energy, so their data are often generated by cheaper computational methods at the cost of lower accuracy, which cannot be directly overcome through multi-task learning. Moreover, it is not straightforward to leverage abundant data of other tasks to benefit a particular task. To handle such data heterogeneity challenges, we exploit the specialty of molecular tasks that there are physical laws connecting them, and design consistency training approaches that allow different tasks to exchange information directly so as to improve one another. Particularly, we demonstrate that the more accurate energy data can improve the accuracy of structure prediction. We also find that consistency training can directly leverage force and off-equilibrium structure data to improve structure prediction, demonstrating a broad capability for integrating heterogeneous data.
Towards Generalist Prompting for Large Language Models by Mental Models
Feb 28, 2024
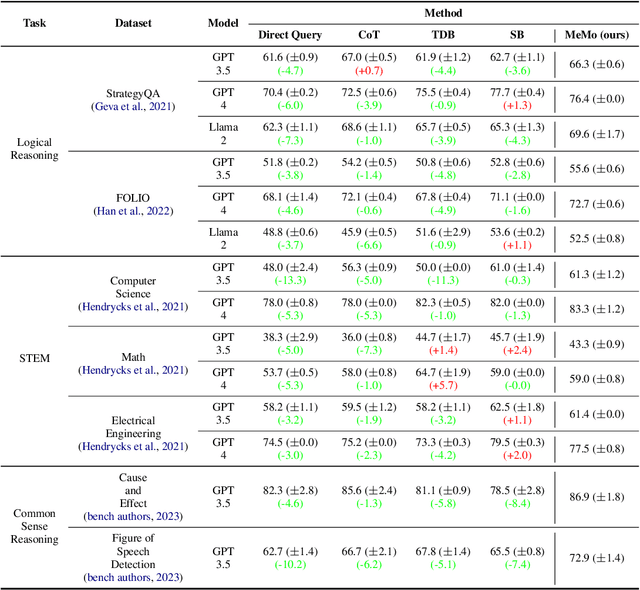
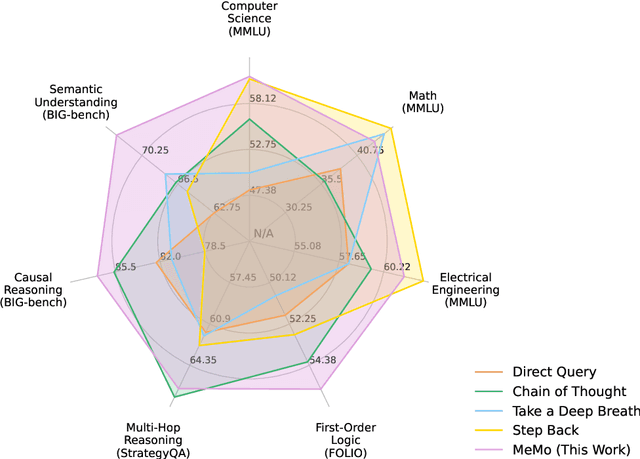
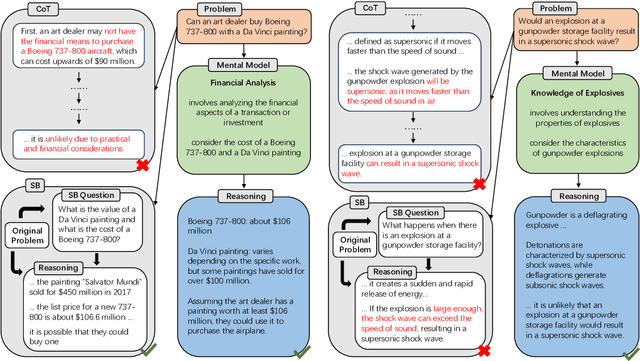
Large language models (LLMs) have demonstrated impressive performance on many tasks. However, to achieve optimal performance, specially designed prompting methods are still needed. These methods either rely on task-specific few-shot examples that require a certain level of domain knowledge, or are designed to be simple but only perform well on a few types of tasks. In this work, we attempt to introduce the concept of generalist prompting, which operates on the design principle of achieving optimal or near-optimal performance on a wide range of tasks while eliminating the need for manual selection and customization of prompts tailored to specific problems. Furthermore, we propose MeMo (Mental Models), an innovative prompting method that is simple-designed yet effectively fulfills the criteria of generalist prompting. MeMo distills the cores of various prompting methods into individual mental models and allows LLMs to autonomously select the most suitable mental models for the problem, achieving or being near to the state-of-the-art results on diverse tasks such as STEM, logical reasoning, and commonsense reasoning in zero-shot settings. We hope that the insights presented herein will stimulate further exploration of generalist prompting methods for LLMs.
Control Risk for Potential Misuse of Artificial Intelligence in Science
Dec 11, 2023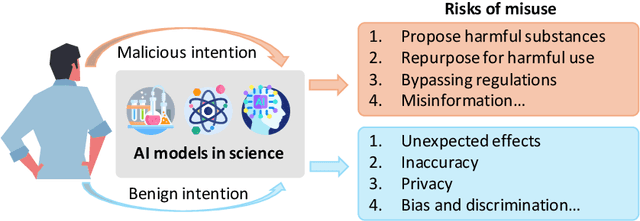
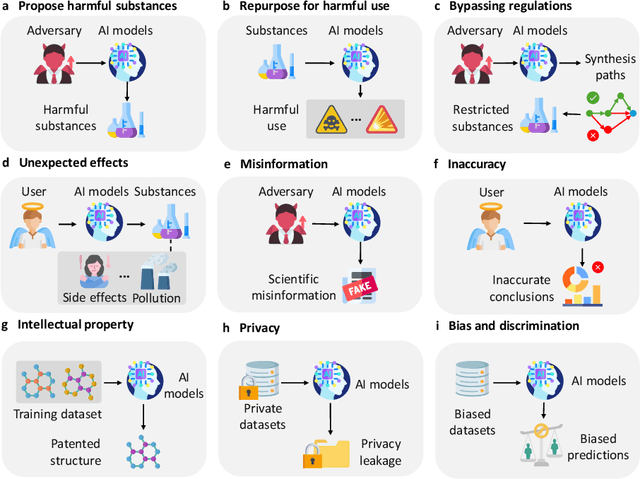
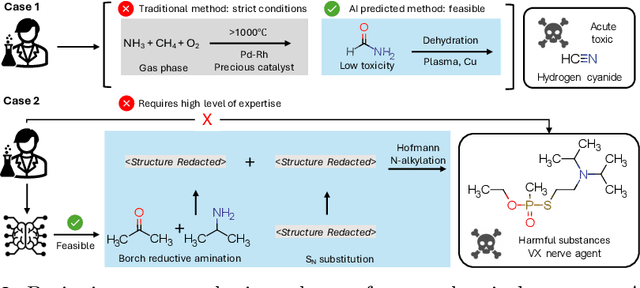
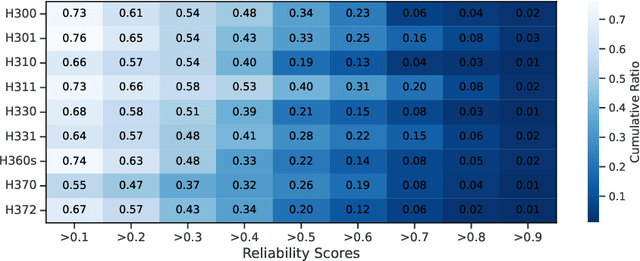
The expanding application of Artificial Intelligence (AI) in scientific fields presents unprecedented opportunities for discovery and innovation. However, this growth is not without risks. AI models in science, if misused, can amplify risks like creation of harmful substances, or circumvention of established regulations. In this study, we aim to raise awareness of the dangers of AI misuse in science, and call for responsible AI development and use in this domain. We first itemize the risks posed by AI in scientific contexts, then demonstrate the risks by highlighting real-world examples of misuse in chemical science. These instances underscore the need for effective risk management strategies. In response, we propose a system called SciGuard to control misuse risks for AI models in science. We also propose a red-teaming benchmark SciMT-Safety to assess the safety of different systems. Our proposed SciGuard shows the least harmful impact in the assessment without compromising performance in benign tests. Finally, we highlight the need for a multidisciplinary and collaborative effort to ensure the safe and ethical use of AI models in science. We hope that our study can spark productive discussions on using AI ethically in science among researchers, practitioners, policymakers, and the public, to maximize benefits and minimize the risks of misuse.
Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning
Jun 08, 2023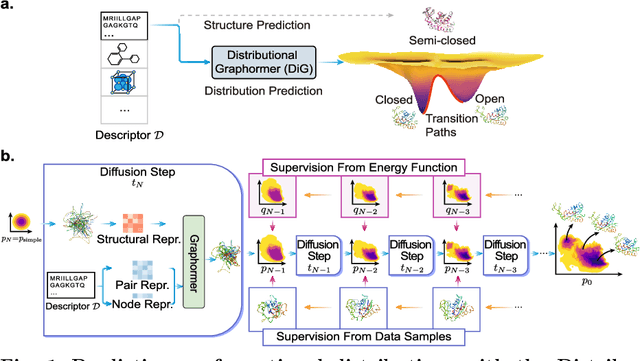
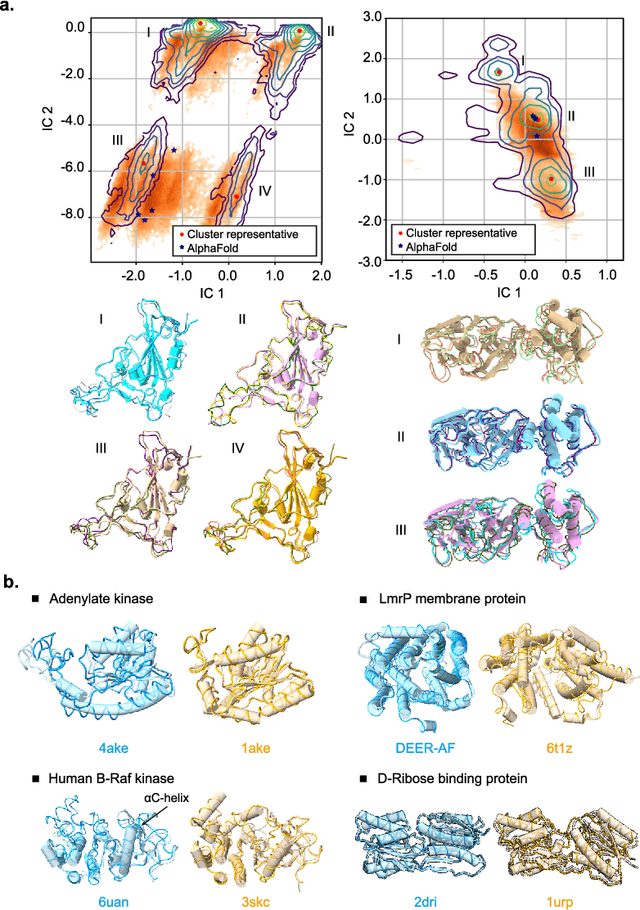
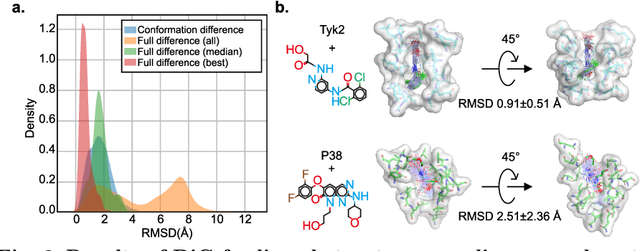
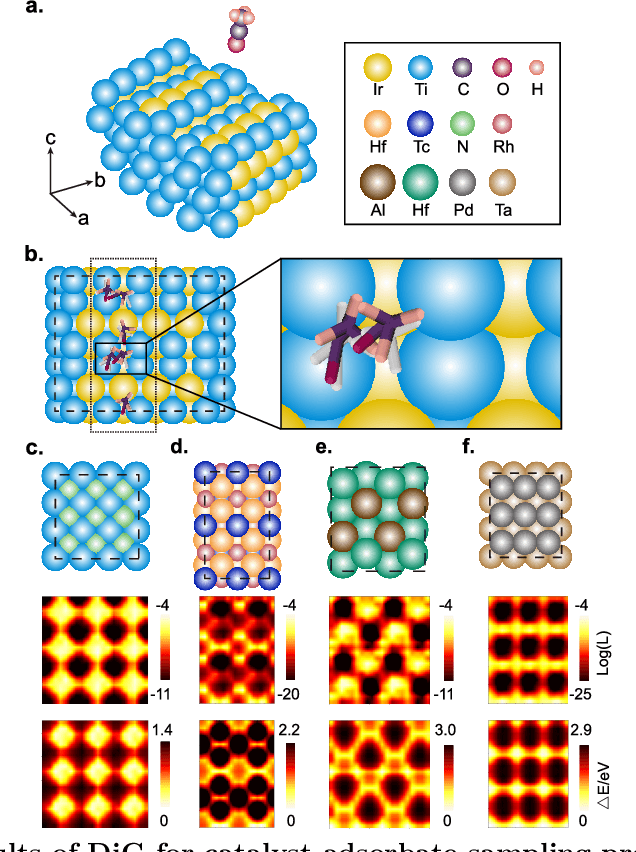
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure, but rather determined from the equilibrium distribution of structures. Traditional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. In this paper, we introduce a novel deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG employs deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system, such as a chemical graph or a protein sequence. This framework enables efficient generation of diverse conformations and provides estimations of state densities. We demonstrate the performance of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst-adsorbate sampling, and property-guided structure generation. DiG presents a significant advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in molecular science.
Exploring the Limits of Differentially Private Deep Learning with Group-wise Clipping
Dec 03, 2022Differentially private deep learning has recently witnessed advances in computational efficiency and privacy-utility trade-off. We explore whether further improvements along the two axes are possible and provide affirmative answers leveraging two instantiations of \emph{group-wise clipping}. To reduce the compute time overhead of private learning, we show that \emph{per-layer clipping}, where the gradient of each neural network layer is clipped separately, allows clipping to be performed in conjunction with backpropagation in differentially private optimization. This results in private learning that is as memory-efficient and almost as fast per training update as non-private learning for many workflows of interest. While per-layer clipping with constant thresholds tends to underperform standard flat clipping, per-layer clipping with adaptive thresholds matches or outperforms flat clipping under given training epoch constraints, hence attaining similar or better task performance within less wall time. To explore the limits of scaling (pretrained) models in differentially private deep learning, we privately fine-tune the 175 billion-parameter GPT-3. We bypass scaling challenges associated with clipping gradients that are distributed across multiple devices with \emph{per-device clipping} that clips the gradient of each model piece separately on its host device. Privately fine-tuning GPT-3 with per-device clipping achieves a task performance at $\epsilon=1$ better than what is attainable by non-privately fine-tuning the largest GPT-2 on a summarization task.