 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUniGenX: Unified Generation of Sequence and Structure with Autoregressive Diffusion
Mar 09, 2025Unified generation of sequence and structure for scientific data (e.g., materials, molecules, proteins) is a critical task. Existing approaches primarily rely on either autoregressive sequence models or diffusion models, each offering distinct advantages and facing notable limitations. Autoregressive models, such as GPT, Llama, and Phi-4, have demonstrated remarkable success in natural language generation and have been extended to multimodal tasks (e.g., image, video, and audio) using advanced encoders like VQ-VAE to represent complex modalities as discrete sequences. However, their direct application to scientific domains is challenging due to the high precision requirements and the diverse nature of scientific data. On the other hand, diffusion models excel at generating high-dimensional scientific data, such as protein, molecule, and material structures, with remarkable accuracy. Yet, their inability to effectively model sequences limits their potential as general-purpose multimodal foundation models. To address these challenges, we propose UniGenX, a unified framework that combines autoregressive next-token prediction with conditional diffusion models. This integration leverages the strengths of autoregressive models to ease the training of conditional diffusion models, while diffusion-based generative heads enhance the precision of autoregressive predictions. We validate the effectiveness of UniGenX on material and small molecule generation tasks, achieving a significant leap in state-of-the-art performance for material crystal structure prediction and establishing new state-of-the-art results for small molecule structure prediction, de novo design, and conditional generation. Notably, UniGenX demonstrates significant improvements, especially in handling long sequences for complex structures, showcasing its efficacy as a versatile tool for scientific data generation.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
Inverse Design of Vitrimeric Polymers by Molecular Dynamics and Generative Modeling
Dec 06, 2023



Vitrimer is a new class of sustainable polymers with the ability of self-healing through rearrangement of dynamic covalent adaptive networks. However, a limited choice of constituent molecules restricts their property space, prohibiting full realization of their potential applications. Through a combination of molecular dynamics (MD) simulations and machine learning (ML), particularly a novel graph variational autoencoder (VAE) model, we establish a method for generating novel vitrimers and guide their inverse design based on desired glass transition temperature (Tg). We build the first vitrimer dataset of one million and calculate Tg on 8,424 of them by high-throughput MD simulations calibrated by a Gaussian process model. The proposed VAE employs dual graph encoders and a latent dimension overlapping scheme which allows for individual representation of multi-component vitrimers. By constructing a continuous latent space containing necessary information of vitrimers, we demonstrate high accuracy and efficiency of our framework in discovering novel vitrimers with desirable Tg beyond the training regime. The proposed vitrimers with reasonable synthesizability cover a wide range of Tg and broaden the potential widespread usage of vitrimeric materials.
BatteryML:An Open-source platform for Machine Learning on Battery Degradation
Oct 23, 2023Battery degradation remains a pivotal concern in the energy storage domain, with machine learning emerging as a potent tool to drive forward insights and solutions. However, this intersection of electrochemical science and machine learning poses complex challenges. Machine learning experts often grapple with the intricacies of battery science, while battery researchers face hurdles in adapting intricate models tailored to specific datasets. Beyond this, a cohesive standard for battery degradation modeling, inclusive of data formats and evaluative benchmarks, is conspicuously absent. Recognizing these impediments, we present BatteryML - a one-step, all-encompass, and open-source platform designed to unify data preprocessing, feature extraction, and the implementation of both traditional and state-of-the-art models. This streamlined approach promises to enhance the practicality and efficiency of research applications. BatteryML seeks to fill this void, fostering an environment where experts from diverse specializations can collaboratively contribute, thus elevating the collective understanding and advancement of battery research.The code for our project is publicly available on GitHub at https://github.com/microsoft/BatteryML.
Learning Intra- and Inter-Cell Differences for Accurate Battery Lifespan Prediction across Diverse Conditions
Oct 11, 2023Battery life prediction holds significant practical value for battery research and development. Currently, many data-driven models rely on early electrical signals from specific target batteries to predict their lifespan. A common shortfall is that most existing methods are developed based on specific aging conditions, which not only limits their model's capability but also diminishes their effectiveness in predicting degradation under varied conditions. As a result, these models often miss out on fully benefiting from the rich historical data available under other conditions. Here, to address above, we introduce an approach that explicitly captures differences between electrical signals of a target battery and a reference battery, irrespective of their materials and aging conditions, to forecast the target battery life. Through this inter-cell difference, we not only enhance the feature space but also pave the way for a universal battery life prediction framework. Remarkably, our model that combines the inter- and intra-cell differences shines across diverse conditions, standing out in its efficiency and accuracy using all accessible datasets. An essential application of our approach is its capability to leverage data from older batteries effectively, enabling newer batteries to capitalize on insights gained from past batteries. This work not only enriches the battery data utilization strategy but also sets the stage for smarter battery management system in the future.
M-OFDFT: Overcoming the Barrier of Orbital-Free Density Functional Theory for Molecular Systems Using Deep Learning
Sep 28, 2023Orbital-free density functional theory (OFDFT) is a quantum chemistry formulation that has a lower cost scaling than the prevailing Kohn-Sham DFT, which is increasingly desired for contemporary molecular research. However, its accuracy is limited by the kinetic energy density functional, which is notoriously hard to approximate for non-periodic molecular systems. In this work, we propose M-OFDFT, an OFDFT approach capable of solving molecular systems using a deep-learning functional model. We build the essential nonlocality into the model, which is made affordable by the concise density representation as expansion coefficients under an atomic basis. With techniques to address unconventional learning challenges therein, M-OFDFT achieves a comparable accuracy with Kohn-Sham DFT on a wide range of molecules untouched by OFDFT before. More attractively, M-OFDFT extrapolates well to molecules much larger than those in training, which unleashes the appealing scaling for studying large molecules including proteins, representing an advancement of the accuracy-efficiency trade-off frontier in quantum chemistry.
Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning
Jun 08, 2023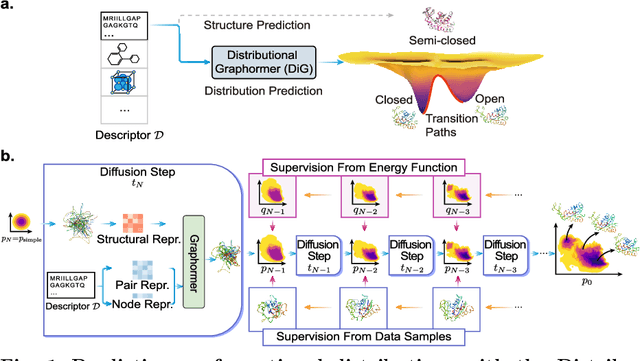
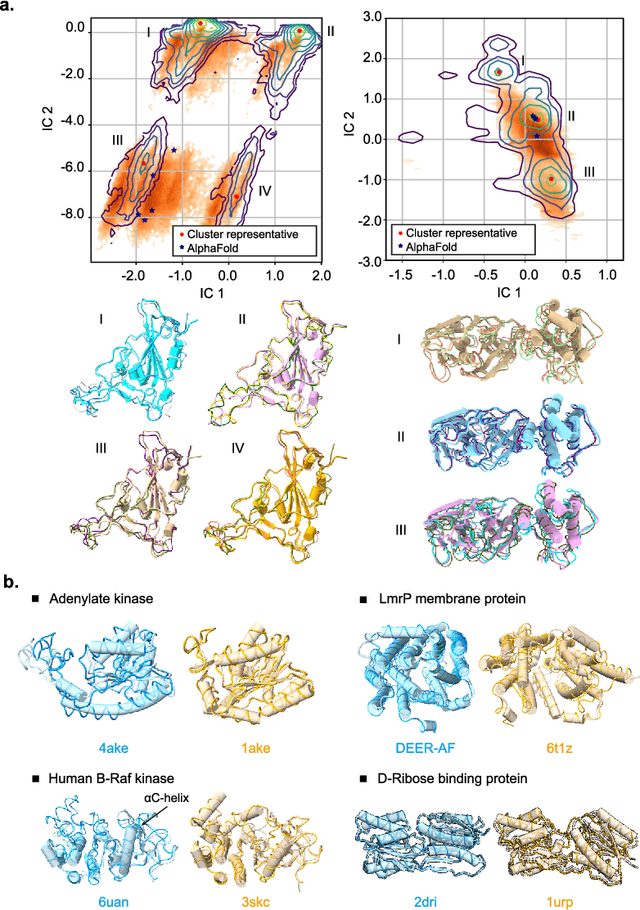
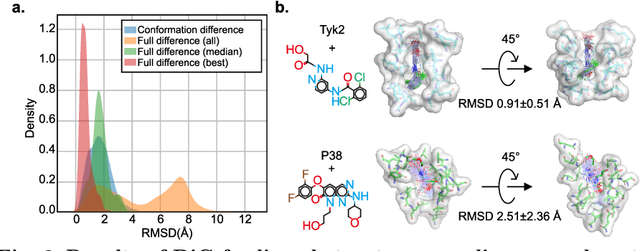
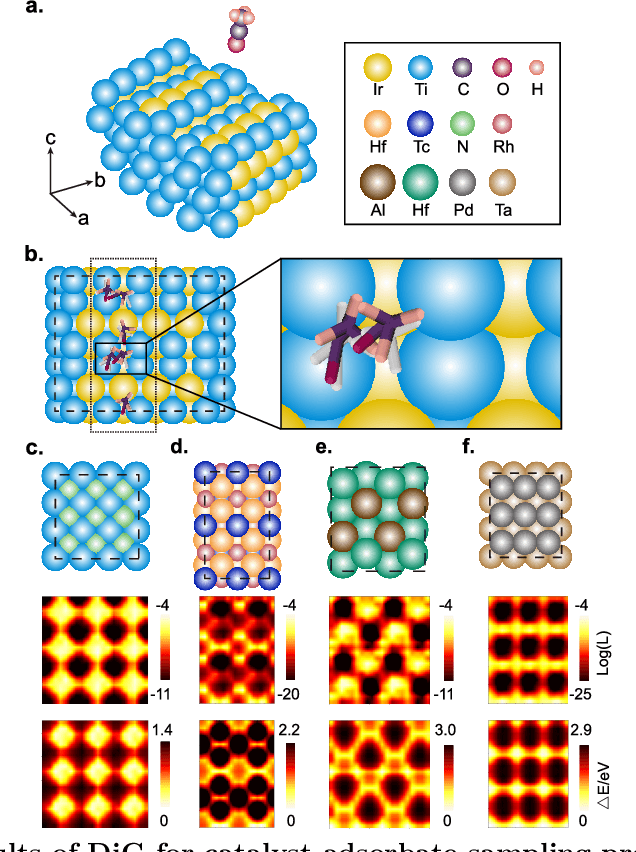
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure, but rather determined from the equilibrium distribution of structures. Traditional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. In this paper, we introduce a novel deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG employs deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system, such as a chemical graph or a protein sequence. This framework enables efficient generation of diverse conformations and provides estimations of state densities. We demonstrate the performance of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst-adsorbate sampling, and property-guided structure generation. DiG presents a significant advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in molecular science.