 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGUIGuard: Toward a General Framework for Privacy-Preserving GUI Agents
Jan 26, 2026GUI agents enable end-to-end automation through direct perception of and interaction with on-screen interfaces. However, these agents frequently access interfaces containing sensitive personal information, and screenshots are often transmitted to remote models, creating substantial privacy risks. These risks are particularly severe in GUI workflows: GUIs expose richer, more accessible private information, and privacy risks depend on interaction trajectories across sequential scenes. We propose GUIGuard, a three-stage framework for privacy-preserving GUI agents: (1) privacy recognition, (2) privacy protection, and (3) task execution under protection. We further construct GUIGuard-Bench, a cross-platform benchmark with 630 trajectories and 13,830 screenshots, annotated with region-level privacy grounding and fine-grained labels of risk level, privacy category, and task necessity. Evaluations reveal that existing agents exhibit limited privacy recognition, with state-of-the-art models achieving only 13.3% accuracy on Android and 1.4% on PC. Under privacy protection, task-planning semantics can still be maintained, with closed-source models showing stronger semantic consistency than open-source ones. Case studies on MobileWorld show that carefully designed protection strategies achieve higher task accuracy while preserving privacy. Our results highlight privacy recognition as a critical bottleneck for practical GUI agents. Project: https://futuresis.github.io/GUIGuard-page/
Population-Evolve: a Parallel Sampling and Evolutionary Method for LLM Math Reasoning
Dec 22, 2025Test-time scaling has emerged as a promising direction for enhancing the reasoning capabilities of Large Language Models in last few years. In this work, we propose Population-Evolve, a training-free method inspired by Genetic Algorithms to optimize LLM reasoning. Our approach maintains a dynamic population of candidate solutions for each problem via parallel reasoning. By incorporating an evolve prompt, the LLM self-evolves its population in all iterations. Upon convergence, the final answer is derived via majority voting. Furthermore, we establish a unification framework that interprets existing test-time scaling strategies through the lens of genetic algorithms. Empirical results demonstrate that Population-Evolve achieves superior accuracy with low performance variance and computational efficiency. Our findings highlight the potential of evolutionary strategies to unlock the reasoning power of LLMs during inference.
Finch: Benchmarking Finance & Accounting across Spreadsheet-Centric Enterprise Workflows
Dec 19, 2025We introduce a finance & accounting benchmark (Finch) for evaluating AI agents on real-world, enterprise-grade professional workflows -- interleaving data entry, structuring, formatting, web search, cross-file retrieval, calculation, modeling, validation, translation, visualization, and reporting. Finch is sourced from authentic enterprise workspaces at Enron (15,000 spreadsheets and 500,000 emails from 150 employees) and other financial institutions, preserving in-the-wild messiness across multimodal artifacts (text, tables, formulas, charts, code, and images) and spanning diverse domains such as budgeting, trading, and asset management. We propose a workflow construction process that combines LLM-assisted discovery with expert annotation: (1) LLM-assisted, expert-verified derivation of workflows from real-world email threads and version histories of spreadsheet files, and (2) meticulous expert annotation for workflows, requiring over 700 hours of domain-expert effort. This yields 172 composite workflows with 384 tasks, involving 1,710 spreadsheets with 27 million cells, along with PDFs and other artifacts, capturing the intrinsically messy, long-horizon, knowledge-intensive, and collaborative nature of real-world enterprise work. We conduct both human and automated evaluations of frontier AI systems including GPT 5.1, Claude Sonnet 4.5, Gemini 3 Pro, Grok 4, and Qwen 3 Max, and GPT 5.1 Pro spends 48 hours in total yet passes only 38.4% of workflows, while Claude Sonnet 4.5 passes just 25.0%. Comprehensive case studies further surface the challenges that real-world enterprise workflows pose for AI agents.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
Bridging Geometric States via Geometric Diffusion Bridge
Oct 31, 2024

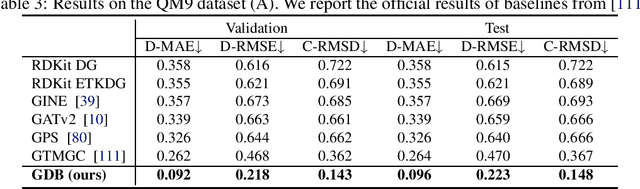
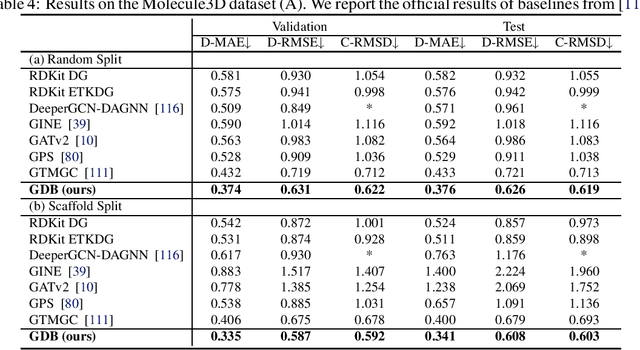
The accurate prediction of geometric state evolution in complex systems is critical for advancing scientific domains such as quantum chemistry and material modeling. Traditional experimental and computational methods face challenges in terms of environmental constraints and computational demands, while current deep learning approaches still fall short in terms of precision and generality. In this work, we introduce the Geometric Diffusion Bridge (GDB), a novel generative modeling framework that accurately bridges initial and target geometric states. GDB leverages a probabilistic approach to evolve geometric state distributions, employing an equivariant diffusion bridge derived by a modified version of Doob's $h$-transform for connecting geometric states. This tailored diffusion process is anchored by initial and target geometric states as fixed endpoints and governed by equivariant transition kernels. Moreover, trajectory data can be seamlessly leveraged in our GDB framework by using a chain of equivariant diffusion bridges, providing a more detailed and accurate characterization of evolution dynamics. Theoretically, we conduct a thorough examination to confirm our framework's ability to preserve joint distributions of geometric states and capability to completely model the underlying dynamics inducing trajectory distributions with negligible error. Experimental evaluations across various real-world scenarios show that GDB surpasses existing state-of-the-art approaches, opening up a new pathway for accurately bridging geometric states and tackling crucial scientific challenges with improved accuracy and applicability.
GeoMFormer: A General Architecture for Geometric Molecular Representation Learning
Jun 24, 2024Molecular modeling, a central topic in quantum mechanics, aims to accurately calculate the properties and simulate the behaviors of molecular systems. The molecular model is governed by physical laws, which impose geometric constraints such as invariance and equivariance to coordinate rotation and translation. While numerous deep learning approaches have been developed to learn molecular representations under these constraints, most of them are built upon heuristic and costly modules. We argue that there is a strong need for a general and flexible framework for learning both invariant and equivariant features. In this work, we introduce a novel Transformer-based molecular model called GeoMFormer to achieve this goal. Using the standard Transformer modules, two separate streams are developed to maintain and learn invariant and equivariant representations. Carefully designed cross-attention modules bridge the two streams, allowing information fusion and enhancing geometric modeling in each stream. As a general and flexible architecture, we show that many previous architectures can be viewed as special instantiations of GeoMFormer. Extensive experiments are conducted to demonstrate the power of GeoMFormer. All empirical results show that GeoMFormer achieves strong performance on both invariant and equivariant tasks of different types and scales. Code and models will be made publicly available at https://github.com/c-tl/GeoMFormer.
Towards Generalist Prompting for Large Language Models by Mental Models
Feb 28, 2024
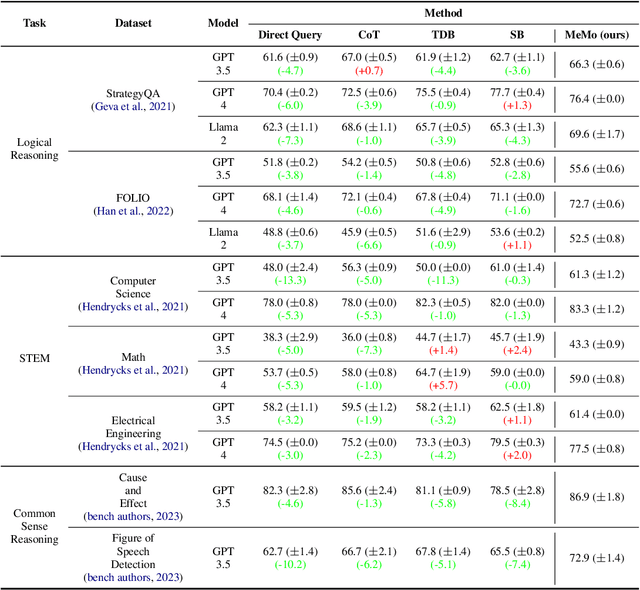
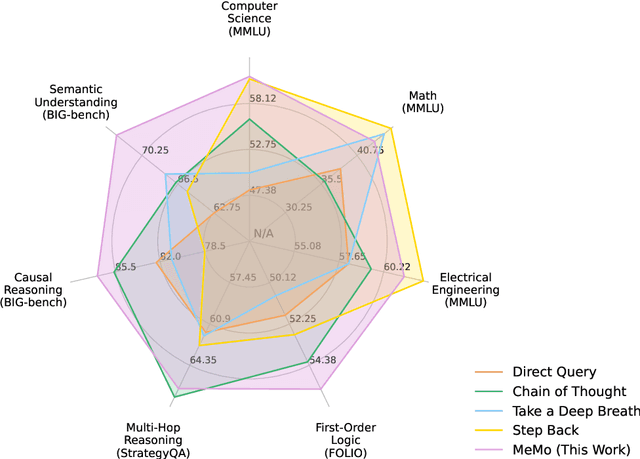
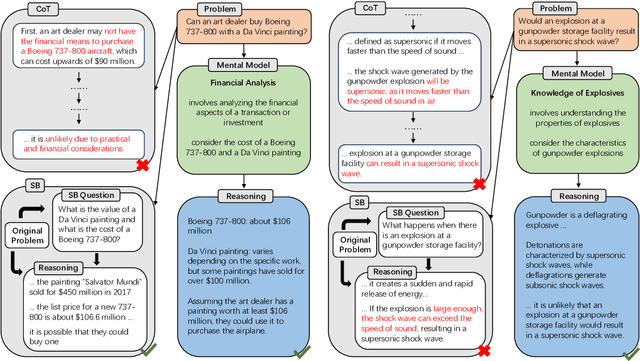
Large language models (LLMs) have demonstrated impressive performance on many tasks. However, to achieve optimal performance, specially designed prompting methods are still needed. These methods either rely on task-specific few-shot examples that require a certain level of domain knowledge, or are designed to be simple but only perform well on a few types of tasks. In this work, we attempt to introduce the concept of generalist prompting, which operates on the design principle of achieving optimal or near-optimal performance on a wide range of tasks while eliminating the need for manual selection and customization of prompts tailored to specific problems. Furthermore, we propose MeMo (Mental Models), an innovative prompting method that is simple-designed yet effectively fulfills the criteria of generalist prompting. MeMo distills the cores of various prompting methods into individual mental models and allows LLMs to autonomously select the most suitable mental models for the problem, achieving or being near to the state-of-the-art results on diverse tasks such as STEM, logical reasoning, and commonsense reasoning in zero-shot settings. We hope that the insights presented herein will stimulate further exploration of generalist prompting methods for LLMs.
Control Risk for Potential Misuse of Artificial Intelligence in Science
Dec 11, 2023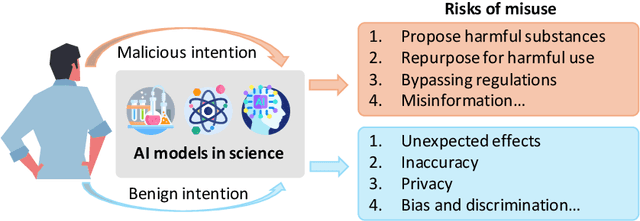
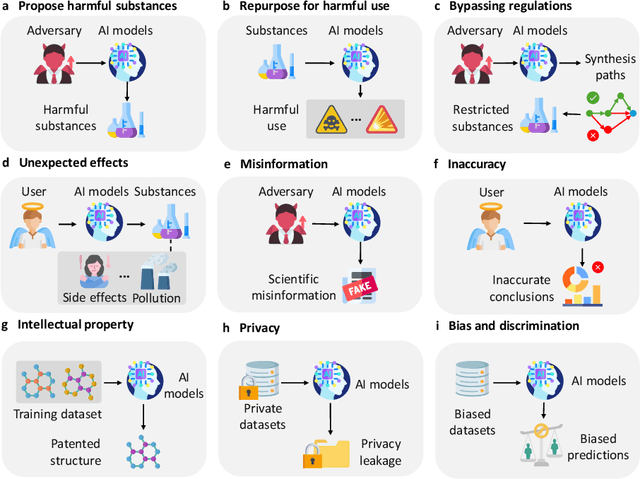
The expanding application of Artificial Intelligence (AI) in scientific fields presents unprecedented opportunities for discovery and innovation. However, this growth is not without risks. AI models in science, if misused, can amplify risks like creation of harmful substances, or circumvention of established regulations. In this study, we aim to raise awareness of the dangers of AI misuse in science, and call for responsible AI development and use in this domain. We first itemize the risks posed by AI in scientific contexts, then demonstrate the risks by highlighting real-world examples of misuse in chemical science. These instances underscore the need for effective risk management strategies. In response, we propose a system called SciGuard to control misuse risks for AI models in science. We also propose a red-teaming benchmark SciMT-Safety to assess the safety of different systems. Our proposed SciGuard shows the least harmful impact in the assessment without compromising performance in benign tests. Finally, we highlight the need for a multidisciplinary and collaborative effort to ensure the safe and ethical use of AI models in science. We hope that our study can spark productive discussions on using AI ethically in science among researchers, practitioners, policymakers, and the public, to maximize benefits and minimize the risks of misuse.
Inverse Design of Vitrimeric Polymers by Molecular Dynamics and Generative Modeling
Dec 06, 2023



Vitrimer is a new class of sustainable polymers with the ability of self-healing through rearrangement of dynamic covalent adaptive networks. However, a limited choice of constituent molecules restricts their property space, prohibiting full realization of their potential applications. Through a combination of molecular dynamics (MD) simulations and machine learning (ML), particularly a novel graph variational autoencoder (VAE) model, we establish a method for generating novel vitrimers and guide their inverse design based on desired glass transition temperature (Tg). We build the first vitrimer dataset of one million and calculate Tg on 8,424 of them by high-throughput MD simulations calibrated by a Gaussian process model. The proposed VAE employs dual graph encoders and a latent dimension overlapping scheme which allows for individual representation of multi-component vitrimers. By constructing a continuous latent space containing necessary information of vitrimers, we demonstrate high accuracy and efficiency of our framework in discovering novel vitrimers with desirable Tg beyond the training regime. The proposed vitrimers with reasonable synthesizability cover a wide range of Tg and broaden the potential widespread usage of vitrimeric materials.
M-OFDFT: Overcoming the Barrier of Orbital-Free Density Functional Theory for Molecular Systems Using Deep Learning
Sep 28, 2023Orbital-free density functional theory (OFDFT) is a quantum chemistry formulation that has a lower cost scaling than the prevailing Kohn-Sham DFT, which is increasingly desired for contemporary molecular research. However, its accuracy is limited by the kinetic energy density functional, which is notoriously hard to approximate for non-periodic molecular systems. In this work, we propose M-OFDFT, an OFDFT approach capable of solving molecular systems using a deep-learning functional model. We build the essential nonlocality into the model, which is made affordable by the concise density representation as expansion coefficients under an atomic basis. With techniques to address unconventional learning challenges therein, M-OFDFT achieves a comparable accuracy with Kohn-Sham DFT on a wide range of molecules untouched by OFDFT before. More attractively, M-OFDFT extrapolates well to molecules much larger than those in training, which unleashes the appealing scaling for studying large molecules including proteins, representing an advancement of the accuracy-efficiency trade-off frontier in quantum chemistry.