 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMIRNet: Integrating Constrained Graph-Based Reasoning with Pre-training for Diagnostic Medical Imaging
Nov 13, 2025Automated interpretation of medical images demands robust modeling of complex visual-semantic relationships while addressing annotation scarcity, label imbalance, and clinical plausibility constraints. We introduce MIRNet (Medical Image Reasoner Network), a novel framework that integrates self-supervised pre-training with constrained graph-based reasoning. Tongue image diagnosis is a particularly challenging domain that requires fine-grained visual and semantic understanding. Our approach leverages self-supervised masked autoencoder (MAE) to learn transferable visual representations from unlabeled data; employs graph attention networks (GAT) to model label correlations through expert-defined structured graphs; enforces clinical priors via constraint-aware optimization using KL divergence and regularization losses; and mitigates imbalance using asymmetric loss (ASL) and boosting ensembles. To address annotation scarcity, we also introduce TongueAtlas-4K, a comprehensive expert-curated benchmark comprising 4,000 images annotated with 22 diagnostic labels--representing the largest public dataset in tongue analysis. Validation shows our method achieves state-of-the-art performance. While optimized for tongue diagnosis, the framework readily generalizes to broader diagnostic medical imaging tasks.
Stabilizing Reinforcement Learning for Honesty Alignment in Language Models on Deductive Reasoning
Nov 12, 2025Reinforcement learning with verifiable rewards (RLVR) has recently emerged as a promising framework for aligning language models with complex reasoning objectives. However, most existing methods optimize only for final task outcomes, leaving models vulnerable to collapse when negative rewards dominate early training. This challenge is especially pronounced in honesty alignment, where models must not only solve answerable queries but also identify when conclusions cannot be drawn from the given premises. Deductive reasoning provides an ideal testbed because it isolates reasoning capability from reliance on external factual knowledge. To investigate honesty alignment, we curate two multi-step deductive reasoning datasets from graph structures, one for linear algebra and one for logical inference, and introduce unanswerable cases by randomly perturbing an edge in half of the instances. We find that GRPO, with or without supervised fine tuning initialization, struggles on these tasks. Through extensive experiments across three models, we evaluate stabilization strategies and show that curriculum learning provides some benefit but requires carefully designed in distribution datasets with controllable difficulty. To address these limitations, we propose Anchor, a reinforcement learning method that injects ground truth trajectories into rollouts, preventing early training collapse. Our results demonstrate that this method stabilizes learning and significantly improves the overall reasoning performance, underscoring the importance of training dynamics for enabling reliable deductive reasoning in aligned language models.
Improving Multislice Electron Ptychography with a Generative Prior
Jul 23, 2025Multislice electron ptychography (MEP) is an inverse imaging technique that computationally reconstructs the highest-resolution images of atomic crystal structures from diffraction patterns. Available algorithms often solve this inverse problem iteratively but are both time consuming and produce suboptimal solutions due to their ill-posed nature. We develop MEP-Diffusion, a diffusion model trained on a large database of crystal structures specifically for MEP to augment existing iterative solvers. MEP-Diffusion is easily integrated as a generative prior into existing reconstruction methods via Diffusion Posterior Sampling (DPS). We find that this hybrid approach greatly enhances the quality of the reconstructed 3D volumes, achieving a 90.50% improvement in SSIM over existing methods.
HeuriGym: An Agentic Benchmark for LLM-Crafted Heuristics in Combinatorial Optimization
Jun 09, 2025While Large Language Models (LLMs) have demonstrated significant advancements in reasoning and agent-based problem-solving, current evaluation methodologies fail to adequately assess their capabilities: existing benchmarks either rely on closed-ended questions prone to saturation and memorization, or subjective comparisons that lack consistency and rigor. In this work, we introduce HeuriGym, an agentic framework designed for evaluating heuristic algorithms generated by LLMs for combinatorial optimization problems, characterized by clearly defined objectives and expansive solution spaces. HeuriGym empowers LLMs to propose heuristics, receive evaluative feedback via code execution, and iteratively refine their solutions. We evaluate nine state-of-the-art models on nine problems across domains such as computer systems, logistics, and biology, exposing persistent limitations in tool use, planning, and adaptive reasoning. To quantify performance, we propose the Quality-Yield Index (QYI), a metric that captures both solution pass rate and quality. Even top models like GPT-o4-mini-high and Gemini-2.5-Pro attain QYI scores of only 0.6, well below the expert baseline of 1. Our open-source benchmark aims to guide the development of LLMs toward more effective and realistic problem-solving in scientific and engineering domains.
Graffe: Graph Representation Learning via Diffusion Probabilistic Models
May 08, 2025Diffusion probabilistic models (DPMs), widely recognized for their potential to generate high-quality samples, tend to go unnoticed in representation learning. While recent progress has highlighted their potential for capturing visual semantics, adapting DPMs to graph representation learning remains in its infancy. In this paper, we introduce Graffe, a self-supervised diffusion model proposed for graph representation learning. It features a graph encoder that distills a source graph into a compact representation, which, in turn, serves as the condition to guide the denoising process of the diffusion decoder. To evaluate the effectiveness of our model, we first explore the theoretical foundations of applying diffusion models to representation learning, proving that the denoising objective implicitly maximizes the conditional mutual information between data and its representation. Specifically, we prove that the negative logarithm of the denoising score matching loss is a tractable lower bound for the conditional mutual information. Empirically, we conduct a series of case studies to validate our theoretical insights. In addition, Graffe delivers competitive results under the linear probing setting on node and graph classification tasks, achieving state-of-the-art performance on 9 of the 11 real-world datasets. These findings indicate that powerful generative models, especially diffusion models, serve as an effective tool for graph representation learning.
Diffusion Variational Inference: Diffusion Models as Expressive Variational Posteriors
Jan 05, 2024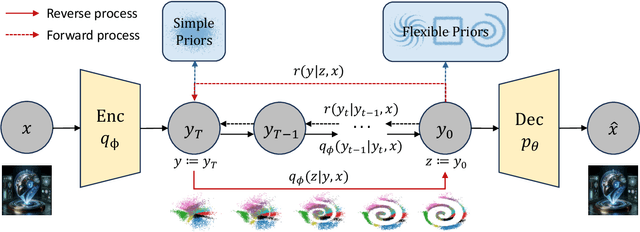
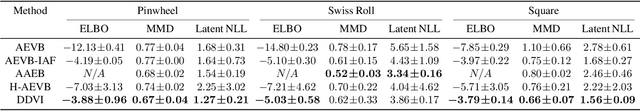
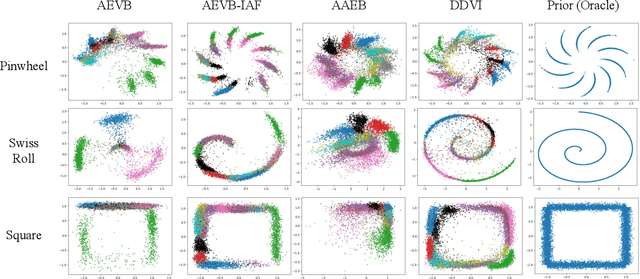
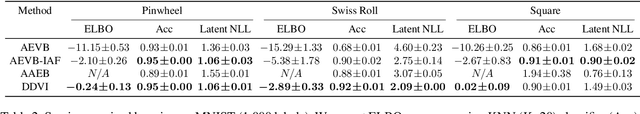
We propose denoising diffusion variational inference (DDVI), an approximate inference algorithm for latent variable models which relies on diffusion models as expressive variational posteriors. Our method augments variational posteriors with auxiliary latents, which yields an expressive class of models that perform diffusion in latent space by reversing a user-specified noising process. We fit these models by optimizing a novel lower bound on the marginal likelihood inspired by the wake-sleep algorithm. Our method is easy to implement (it fits a regularized extension of the ELBO), is compatible with black-box variational inference, and outperforms alternative classes of approximate posteriors based on normalizing flows or adversarial networks. When applied to deep latent variable models, our method yields the denoising diffusion VAE (DD-VAE) algorithm. We use this algorithm on a motivating task in biology -- inferring latent ancestry from human genomes -- outperforming strong baselines on the Thousand Genomes dataset.
ModuLoRA: Finetuning 3-Bit LLMs on Consumer GPUs by Integrating with Modular Quantizers
Sep 28, 2023



We propose a memory-efficient finetuning algorithm for large language models (LLMs) that supports finetuning LLMs with 65B parameters in 3-bit or 4-bit precision on as little as one 48GB GPU. Our method, modular low-rank adaptation (ModuLoRA), integrates any user-specified weight quantizer with finetuning via low-rank adapters (LoRAs). Our approach relies on a simple quantization-agnostic backward pass that adaptively materializes low-precision LLM weights from a custom black-box quantization module. This approach enables finetuning 3-bit LLMs for the first time--leveraging state-of-the-art 3-bit OPTQ quantization often outperforms finetuning that relies on less sophisticated 4-bit and 8-bit methods. In our experiments, ModuLoRA attains competitive performance on text classification, natural language infernece, and instruction following tasks using significantly less memory than existing approaches, and we also surpass the state-of-the-art ROUGE score on a popular summarization task. We release ModuLoRA together with a series of low-precision models--including the first family of 3-bit instruction following Alpaca LLMs--as part of LLMTOOLS, a user-friendly library for quantizing, running, and finetuning LLMs on consumer GPUs.
InfoDiffusion: Representation Learning Using Information Maximizing Diffusion Models
Jun 14, 2023



While diffusion models excel at generating high-quality samples, their latent variables typically lack semantic meaning and are not suitable for representation learning. Here, we propose InfoDiffusion, an algorithm that augments diffusion models with low-dimensional latent variables that capture high-level factors of variation in the data. InfoDiffusion relies on a learning objective regularized with the mutual information between observed and hidden variables, which improves latent space quality and prevents the latents from being ignored by expressive diffusion-based decoders. Empirically, we find that InfoDiffusion learns disentangled and human-interpretable latent representations that are competitive with state-of-the-art generative and contrastive methods, while retaining the high sample quality of diffusion models. Our method enables manipulating the attributes of generated images and has the potential to assist tasks that require exploring a learned latent space to generate quality samples, e.g., generative design.
Time Series Contrastive Learning with Information-Aware Augmentations
Mar 21, 2023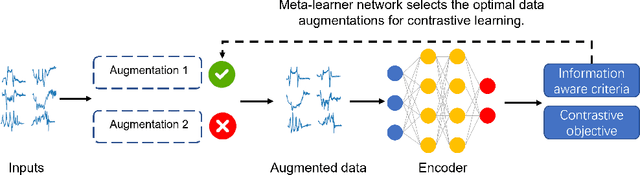
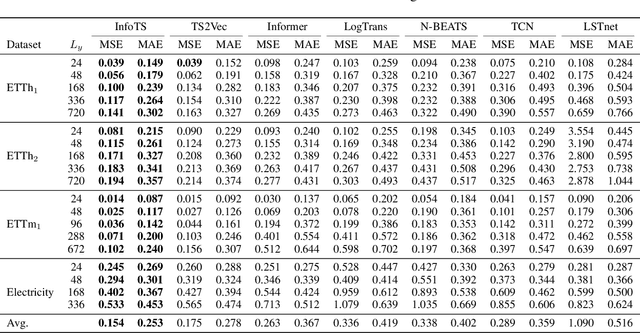
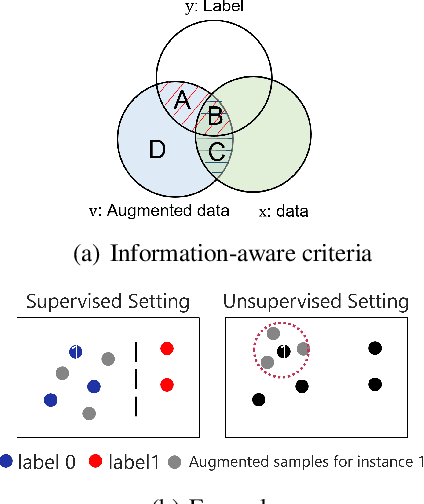
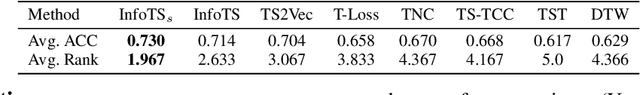
Various contrastive learning approaches have been proposed in recent years and achieve significant empirical success. While effective and prevalent, contrastive learning has been less explored for time series data. A key component of contrastive learning is to select appropriate augmentations imposing some priors to construct feasible positive samples, such that an encoder can be trained to learn robust and discriminative representations. Unlike image and language domains where ``desired'' augmented samples can be generated with the rule of thumb guided by prefabricated human priors, the ad-hoc manual selection of time series augmentations is hindered by their diverse and human-unrecognizable temporal structures. How to find the desired augmentations of time series data that are meaningful for given contrastive learning tasks and datasets remains an open question. In this work, we address the problem by encouraging both high \textit{fidelity} and \textit{variety} based upon information theory. A theoretical analysis leads to the criteria for selecting feasible data augmentations. On top of that, we propose a new contrastive learning approach with information-aware augmentations, InfoTS, that adaptively selects optimal augmentations for time series representation learning. Experiments on various datasets show highly competitive performance with up to 12.0\% reduction in MSE on forecasting tasks and up to 3.7\% relative improvement in accuracy on classification tasks over the leading baselines.
Xtal2DoS: Attention-based Crystal to Sequence Learning for Density of States Prediction
Feb 03, 2023Modern machine learning techniques have been extensively applied to materials science, especially for property prediction tasks. A majority of these methods address scalar property predictions, while more challenging spectral properties remain less emphasized. We formulate a crystal-to-sequence learning task and propose a novel attention-based learning method, Xtal2DoS, which decodes the sequential representation of the material density of states (DoS) properties by incorporating the learned atomic embeddings through attention networks. Experiments show Xtal2DoS is faster than the existing models, and consistently outperforms other state-of-the-art methods on four metrics for two fundamental spectral properties, phonon and electronic DoS.