 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting the protein-ligand affinity from molecular dynamics trajectories
Aug 19, 2022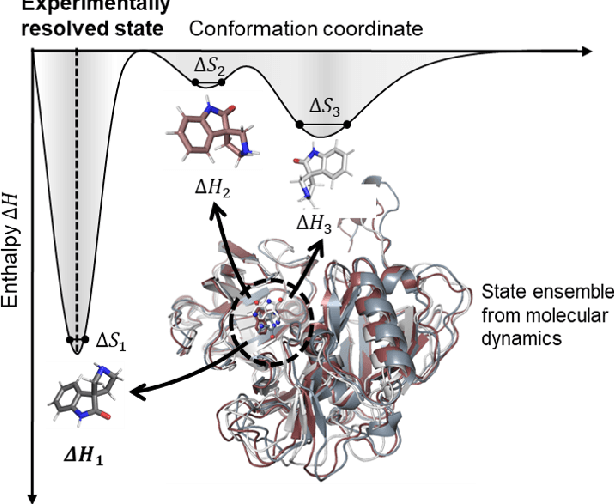
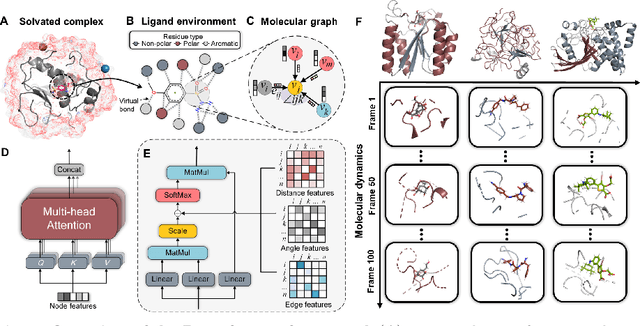
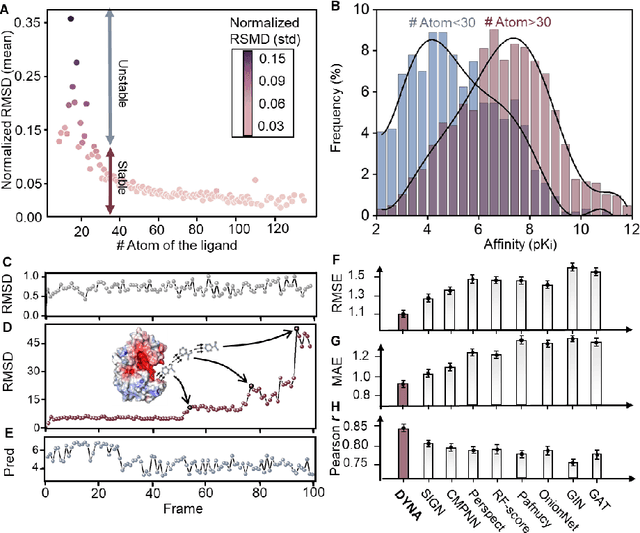
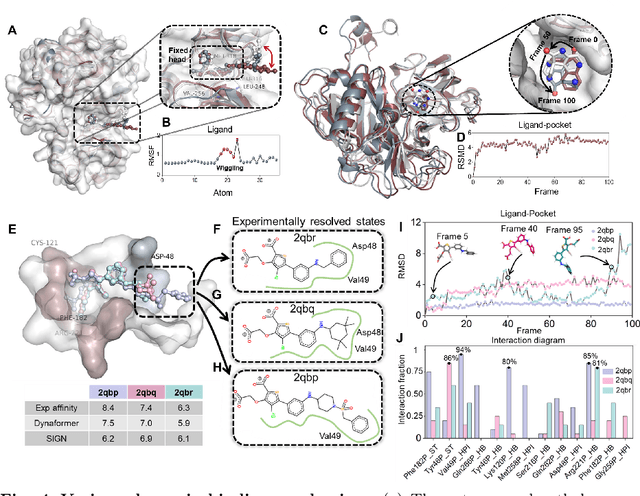
The accurate protein-ligand binding affinity prediction is essential in drug design and many other molecular recognition problems. Despite many advances in affinity prediction based on machine learning techniques, they are still limited since the protein-ligand binding is determined by the dynamics of atoms and molecules. To this end, we curated an MD dataset containing 3,218 dynamic protein-ligand complexes and further developed Dynaformer, a graph-based deep learning framework. Dynaformer can fully capture the dynamic binding rules by considering various geometric characteristics of the interaction. Our method shows superior performance over the methods hitherto reported. Moreover, we performed virtual screening on heat shock protein 90 (HSP90) by integrating our model with structure-based docking. We benchmarked our performance against other baselines, demonstrating that our method can identify the molecule with the highest experimental potency. We anticipate that large-scale MD dataset and machine learning models will form a new synergy, providing a new route towards accelerated drug discovery and optimization.