 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Multi-modal Agentic Co-pilot for Evidence Grounded Computational Pathology
Jun 06, 2026Pathology is the cornerstone of modern medicine, where accurate decision-making relies heavily on evidence-based practices. While artificial intelligence (AI) has the potential to transform clinical workflows, the intersection of AI and evidence-based medicine remains under-explored, with primitive attempts restricted to text-only general medicine. In this work, we present PathPocket, a multimodal AI agentic co-pilot designed specifically for evidence grounded pathology. We construct the most comprehensive pathology evidence corpus to date, encompassing approximately 110,472 public and authorized documents structured across a rigorous hierarchy of evidence from clinical guideline to expert opinion. From this meticulously graded foundation, we build a large-scale multimodal pathology hypergraph containing over 4.55 million entities and 7.10 million relations. Serving as a robust knowledge engine, this hypergraph provides traceable evidence for a collaborative multi-agent reasoning framework integrating input understanding, evidence retrieval, filtering, and diagnosis generation. This enables PathPocket to seamlessly resolve a wide spectrum of clinical tasks, ranging from text-only queries to complex multimodal diagnostics involving region-of-interest (ROI) and gigapixel whole-slide images (WSIs). We rigorously evaluate the system on a multidimensional benchmark of over 200,000 real-world cases, where it significantly outperforms existing state-of-the-arts. Crucially, extensive user studies demonstrate that PathPocket substantially improves the diagnostic accuracy and confidence of pathologists. By directly grounding pathology interpretations in verifiable literature, PathPocket offers a practical and scalable solution for the future of evidence grounded computational pathology.
A Pathology Foundation Model for Gastric Cancer with Real-World Validation
Jun 03, 2026Gastric cancer remains a major cause of cancer mortality, yet its histological and molecular heterogeneity complicates diagnosis and risk stratification. General-purpose pathology foundation models (PFMs) often plateau on fine-grained endpoints central to gastric cancer care, and few have undergone rigorous prospective validation or clinical reader studies. We present GRACE, a Gastric-specific foundation model for Real-world Assessment and Clinical dEcision support. GRACE was developed from multicenter gastric pathology datasets totaling 48,364 primarily HE-stained whole-slide images from 37,493 patients. When evaluated on 28 clinically relevant tasks, GRACE consistently outperformed representative pancancer PFMs, achieving a macro-AUC of 0.9188, with strong performance for precancerous lesion diagnosis (macro-AUC 0.9322), tumor histopathological assessment (macro-AUC 0.9119), molecular profiling (macro-AUC 0.8682), and prognostic prediction. Beyond benchmarking, GRACE's translational value was substantiated through a rigorous evidence chain. Under safety-gated criteria requiring 100% NPV for rule-out and 100% PPV for rule-in, GRACE streamlined review for up to 69.6% of malignancy-diagnosis cases and triaged 46.8% of MMR-IHC follow-up requests. This translational feasibility was further strengthened by a randomized crossover reader study of pathologist-AI collaboration. With GRACE assistance, diagnostic accuracy improved from 82.0% to 89.9%, yielding nearly twofold higher adjusted odds of a correct diagnosis (OR 1.987) alongside concurrent gains in sensitivity and specificity. AI assistance also reduced diagnostic time by 14.9%, elevated diagnostic confidence by 9.0%, and markedly improved inter-rater agreement. When calibrated to maintain non-inferior performance to senior pathologists, the AI-assisted workflow could triage 60.7% of atrophy and 82.7% of intestinal metaplasia cases.
Edge-aware Decoding for Neural Asymmetric Routing
Jun 01, 2026Neural asymmetric routing models increasingly encode directionality through matrix representations and asymmetry-aware attention. The final routing action, however, is not a node in isolation but a directed transition chosen under the current partial route. This creates a representation--decision mismatch: pairwise cost information may be encoded upstream while the final candidate logit is still largely parameterized as context--node compatibility. We propose a decoder-design principle for neural asymmetric routing: the final score should explicitly expose transition-level quantities suggested by the problem's cost-to-go structure. We instantiate this principle with an edge-aware decoder that adds candidate-specific terms for the current directed edge, return-to-start closure, and static lightweight lookahead, while keeping the representation backbone fixed. On a controlled SVD/Sinkhorn asymmetric backbone, the decoder improves over the RADAR reference when trained on ATSP-100 and evaluated zero-shot on ATSP-100/200/500/1000, reducing the ATSP-1000 gap from $4.13\%$ to $2.73\%$. On ACVRP, the same score-level modification shows the same qualitative trend under a richer routing state. ATSP ablations and directed-transition diagnostics sharpen the mechanism: the strongest evidence concerns sensitivity to the current directed edge, while closure and static lookahead act as heuristic continuation cues. The results support a mechanism study: a key decoder-side signal in neural asymmetric routing is decision-time exposure of transition-level edge information.
Spatial Transcriptomics-Guided Alignment Enhances Molecular Profiling in Pathology Foundation Model
May 29, 2026Comprehensive molecular profiling is essential for modern precision oncology but remains hindered by prohibitive costs, specimen exhaustion, and protracted turnaround times. While pathology foundation models (PFMs) have demonstrated potential for inferring molecular phenotypes from routine hematoxylin and eosin (H&E) whole-slide images (WSIs), current architectures primarily rely on vision-centric self-supervised learning or vision-language alignment, lacking the spatially resolved molecular supervision required to connect subtle morphological features with underlying genomic alterations. Spatial transcriptomics (ST) emerges as a transformative technology that enables transcriptomic quantification within intact tissue sections, thereby preserving the precise spatial link between histology and molecular profiles. In this study, we present a Spatial Transcriptomics-guided Alignment framework for Molecular Profiling (STAMP), which endows PFMs with intrinsic molecular awareness. To support this paradigm, we curated HumanST-1k, a human ST dataset spanning diverse anatomical organs and sequencing platforms. This atlas yields 1.8 million pairs of H&E patches and corresponding transcriptomic profiles, providing a corpus that links histological structures with their molecular states. To mitigate the technical noise inherent to raw transcriptomics, STAMP applies a pathway-informed alignment strategy that aggregates transcriptomic data into biologically functional pathways, which are subsequently integrated into PFMs via parameter-efficient fine-tuning. This alignment enriches the representation space of PFMs and unlocks their capacity to resolve sub-visual molecular signatures. The clinical utility of these augmented representations was validated through a multi-tier evaluation framework.
A Clinically Validated Foundation Model for Comprehensive Lung Pathology Interpretation
May 25, 2026Pathological assessment guides lung cancer diagnosis, treatment selection, and prognostic evaluation, yet current CPath approaches rely on task-specific models for isolated objectives. Although pan-cancer foundation models offer versatility, they lack subspecialty-level depth and have not been evaluated across clinical workflows or prospectively validated in real-world settings. We introduce PulmoFoundation, a multi-center, prospectively validated, randomized controlled trial (RCT)-evaluated foundation model for comprehensive lung pathology assessment across pre-operative, intra-operative, and post-operative care. Built upon Virchow2 via subspecialty-specific pretraining using ~40,000 diagnostic H&E-stained whole-slide images (WSIs), PulmoFoundation was systematically evaluated on ~26,000 WSIs across 32 clinically relevant tasks. In addition to accurately predicting molecular markers and patient survival, our model achieves clinical-grade performance in core diagnostic tasks across biopsy, frozen section, and surgical resection slides. In a registered prospective study of 1,357 patients across 11 diagnostic tasks, our model achieved an average AUC of 92.3%. Using pre-specified triage thresholds, PulmoFoundation could reduce additional second-review burden for 68.8% of biopsies and 83.0% of frozen sections, and defer 44.5% of IHC stain orders, with PPVs of 1.0, 0.991, and 0.966. Beyond prospective validation, we conducted a crossover RCT with eight pathologists, in which AI assistance improved diagnostic accuracy across 4,928 case-reader pairs (91.7% w/ AI vs. 83.8% w/o AI). AI assistance also reduced median diagnostic time by 19.6%, increased diagnostic confidence by 8.7%, and improved inter-rater agreement from moderate (kappa = 0.56) to substantial (kappa = 0.76). Together, these evaluations support PulmoFoundation as a clinically validated decision-support system for lung pathology.
A Deployment-Friendly Foundational Framework for Efficient Computational Pathology
Feb 15, 2026Pathology foundation models (PFMs) have enabled robust generalization in computational pathology through large-scale datasets and expansive architectures, but their substantial computational cost, particularly for gigapixel whole slide images, limits clinical accessibility and scalability. Here, we present LitePath, a deployment-friendly foundational framework designed to mitigate model over-parameterization and patch level redundancy. LitePath integrates LiteFM, a compact model distilled from three large PFMs (Virchow2, H-Optimus-1 and UNI2) using 190 million patches, and the Adaptive Patch Selector (APS), a lightweight component for task-specific patch selection. The framework reduces model parameters by 28x and lowers FLOPs by 403.5x relative to Virchow2, enabling deployment on low-power edge hardware such as the NVIDIA Jetson Orin Nano Super. On this device, LitePath processes 208 slides per hour, 104.5x faster than Virchow2, and consumes 0.36 kWh per 3,000 slides, 171x lower than Virchow2 on an RTX3090 GPU. We validated accuracy using 37 cohorts across four organs and 26 tasks (26 internal, 9 external, and 2 prospective), comprising 15,672 slides from 9,808 patients disjoint from the pretraining data. LitePath ranks second among 19 evaluated models and outperforms larger models including H-Optimus-1, mSTAR, UNI2 and GPFM, while retaining 99.71% of the AUC of Virchow2 on average. To quantify the balance between accuracy and efficiency, we propose the Deployability Score (D-Score), defined as the weighted geometric mean of normalized AUC and normalized FLOP, where LitePath achieves the highest value, surpassing Virchow2 by 10.64%. These results demonstrate that LitePath enables rapid, cost-effective and energy-efficient pathology image analysis on accessible hardware while maintaining accuracy comparable to state-of-the-art PFMs and reducing the carbon footprint of AI deployment.
HippMetric: A skeletal-representation-based framework for cross-sectional and longitudinal hippocampal substructural morphometry
Dec 22, 2025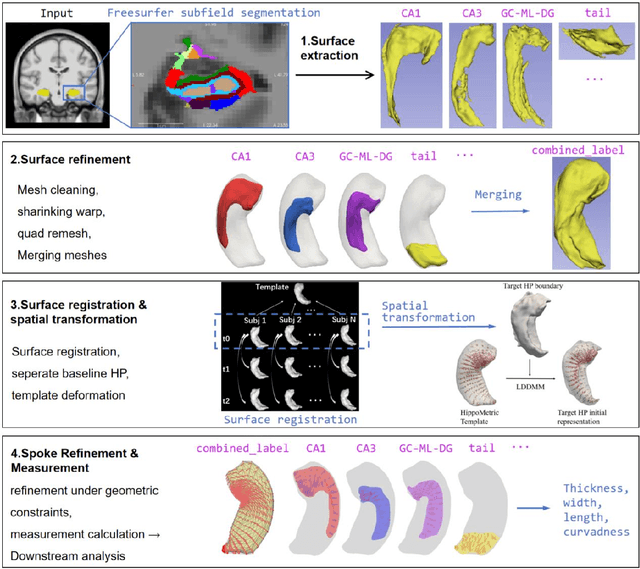
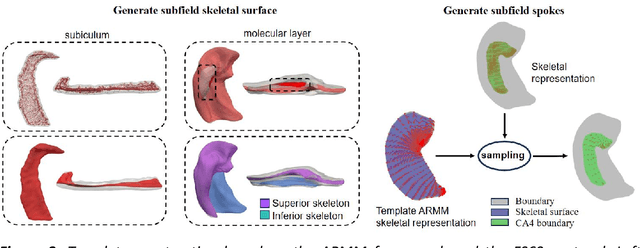
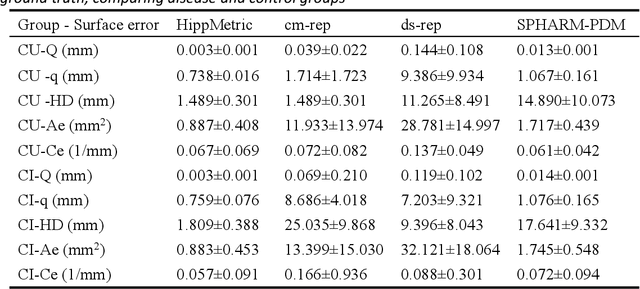
Accurate characterization of hippocampal substructure is crucial for detecting subtle structural changes and identifying early neurodegenerative biomarkers. However, high inter-subject variability and complex folding pattern of human hippocampus hinder consistent cross-subject and longitudinal analysis. Most existing approaches rely on subject-specific modelling and lack a stable intrinsic coordinate system to accommodate anatomical variability, which limits their ability to establish reliable inter- and intra-individual correspondence. To address this, we propose HippMetric, a skeletal representation (s-rep)-based framework for hippocampal substructural morphometry and point-wise correspondence across individuals and scans. HippMetric builds on the Axis-Referenced Morphometric Model (ARMM) and employs a deformable skeletal coordinate system aligned with hippocampal anatomy and function, providing a biologically grounded reference for correspondence. Our framework comprises two core modules: a skeletal-based coordinate system that respects the hippocampus' conserved longitudinal lamellar architecture, in which functional units (lamellae) are stacked perpendicular to the long-axis, enabling anatomically consistent localization across subjects and time; and individualized s-reps generated through surface reconstruction, deformation, and geometrically constrained spoke refinement, enforcing boundary adherence, orthogonality and non-intersection to produce mathematically valid skeletal geometry. Extensive experiments on two international cohorts demonstrate that HippMetric achieves higher accuracy, reliability, and correspondence stability compared to existing shape models.
MambaMIL+: Modeling Long-Term Contextual Patterns for Gigapixel Whole Slide Image
Dec 19, 2025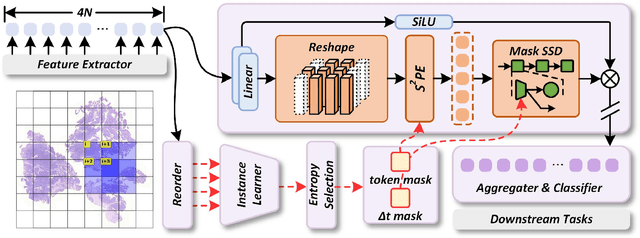
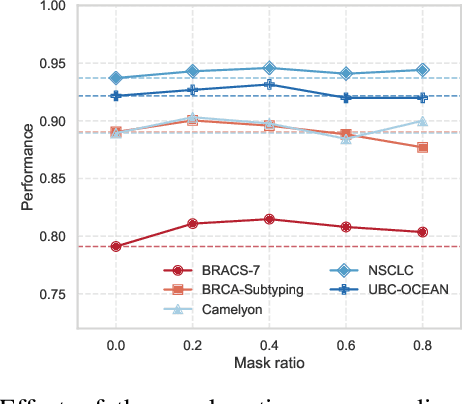
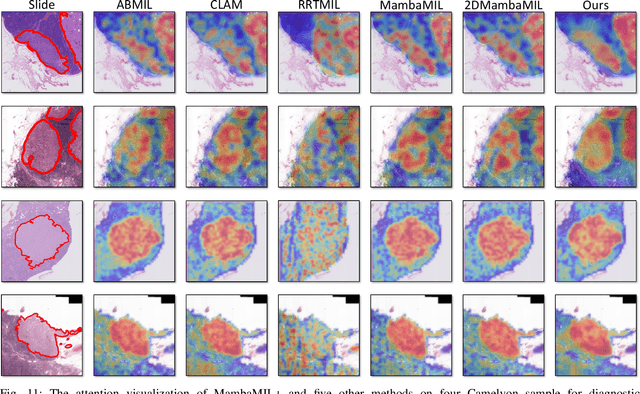
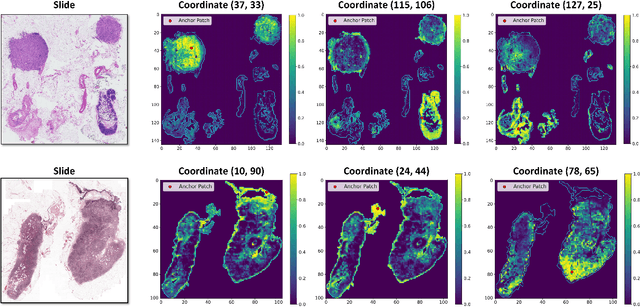
Whole-slide images (WSIs) are an important data modality in computational pathology, yet their gigapixel resolution and lack of fine-grained annotations challenge conventional deep learning models. Multiple instance learning (MIL) offers a solution by treating each WSI as a bag of patch-level instances, but effectively modeling ultra-long sequences with rich spatial context remains difficult. Recently, Mamba has emerged as a promising alternative for long sequence learning, scaling linearly to thousands of tokens. However, despite its efficiency, it still suffers from limited spatial context modeling and memory decay, constraining its effectiveness to WSI analysis. To address these limitations, we propose MambaMIL+, a new MIL framework that explicitly integrates spatial context while maintaining long-range dependency modeling without memory forgetting. Specifically, MambaMIL+ introduces 1) overlapping scanning, which restructures the patch sequence to embed spatial continuity and instance correlations; 2) a selective stripe position encoder (S2PE) that encodes positional information while mitigating the biases of fixed scanning orders; and 3) a contextual token selection (CTS) mechanism, which leverages supervisory knowledge to dynamically enlarge the contextual memory for stable long-range modeling. Extensive experiments on 20 benchmarks across diagnostic classification, molecular prediction, and survival analysis demonstrate that MambaMIL+ consistently achieves state-of-the-art performance under three feature extractors (ResNet-50, PLIP, and CONCH), highlighting its effectiveness and robustness for large-scale computational pathology
Enhancing WSI-Based Survival Analysis with Report-Auxiliary Self-Distillation
Sep 19, 2025Survival analysis based on Whole Slide Images (WSIs) is crucial for evaluating cancer prognosis, as they offer detailed microscopic information essential for predicting patient outcomes. However, traditional WSI-based survival analysis usually faces noisy features and limited data accessibility, hindering their ability to capture critical prognostic features effectively. Although pathology reports provide rich patient-specific information that could assist analysis, their potential to enhance WSI-based survival analysis remains largely unexplored. To this end, this paper proposes a novel Report-auxiliary self-distillation (Rasa) framework for WSI-based survival analysis. First, advanced large language models (LLMs) are utilized to extract fine-grained, WSI-relevant textual descriptions from original noisy pathology reports via a carefully designed task prompt. Next, a self-distillation-based pipeline is designed to filter out irrelevant or redundant WSI features for the student model under the guidance of the teacher model's textual knowledge. Finally, a risk-aware mix-up strategy is incorporated during the training of the student model to enhance both the quantity and diversity of the training data. Extensive experiments carried out on our collected data (CRC) and public data (TCGA-BRCA) demonstrate the superior effectiveness of Rasa against state-of-the-art methods. Our code is available at https://github.com/zhengwang9/Rasa.
Generative AI for Misalignment-Resistant Virtual Staining to Accelerate Histopathology Workflows
Sep 17, 2025Accurate histopathological diagnosis often requires multiple differently stained tissue sections, a process that is time-consuming, labor-intensive, and environmentally taxing due to the use of multiple chemical stains. Recently, virtual staining has emerged as a promising alternative that is faster, tissue-conserving, and environmentally friendly. However, existing virtual staining methods face significant challenges in clinical applications, primarily due to their reliance on well-aligned paired data. Obtaining such data is inherently difficult because chemical staining processes can distort tissue structures, and a single tissue section cannot undergo multiple staining procedures without damage or loss of information. As a result, most available virtual staining datasets are either unpaired or roughly paired, making it difficult for existing methods to achieve accurate pixel-level supervision. To address this challenge, we propose a robust virtual staining framework featuring cascaded registration mechanisms to resolve spatial mismatches between generated outputs and their corresponding ground truth. Experimental results demonstrate that our method significantly outperforms state-of-the-art models across five datasets, achieving an average improvement of 3.2% on internal datasets and 10.1% on external datasets. Moreover, in datasets with substantial misalignment, our approach achieves a remarkable 23.8% improvement in peak signal-to-noise ratio compared to baseline models. The exceptional robustness of the proposed method across diverse datasets simplifies the data acquisition process for virtual staining and offers new insights for advancing its development.