 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLower Ricci Curvature for Hypergraphs
Jun 04, 2025Networks with higher-order interactions, prevalent in biological, social, and information systems, are naturally represented as hypergraphs, yet their structural complexity poses fundamental challenges for geometric characterization. While curvature-based methods offer powerful insights in graph analysis, existing extensions to hypergraphs suffer from critical trade-offs: combinatorial approaches such as Forman-Ricci curvature capture only coarse features, whereas geometric methods like Ollivier-Ricci curvature offer richer expressivity but demand costly optimal transport computations. To address these challenges, we introduce hypergraph lower Ricci curvature (HLRC), a novel curvature metric defined in closed form that achieves a principled balance between interpretability and efficiency. Evaluated across diverse synthetic and real-world hypergraph datasets, HLRC consistently reveals meaningful higher-order organization, distinguishing intra- from inter-community hyperedges, uncovering latent semantic labels, tracking temporal dynamics, and supporting robust clustering of hypergraphs based on global structure. By unifying geometric sensitivity with algorithmic simplicity, HLRC provides a versatile foundation for hypergraph analytics, with broad implications for tasks including node classification, anomaly detection, and generative modeling in complex systems.
Pangu Pro MoE: Mixture of Grouped Experts for Efficient Sparsity
May 28, 2025The surgence of Mixture of Experts (MoE) in Large Language Models promises a small price of execution cost for a much larger model parameter count and learning capacity, because only a small fraction of parameters are activated for each input token. However, it is commonly observed that some experts are activated far more often than others, leading to system inefficiency when running the experts on different devices in parallel. Therefore, we introduce Mixture of Grouped Experts (MoGE), which groups the experts during selection and balances the expert workload better than MoE in nature. It constrains tokens to activate an equal number of experts within each predefined expert group. When a model execution is distributed on multiple devices, this architectural design ensures a balanced computational load across devices, significantly enhancing throughput, particularly for the inference phase. Further, we build Pangu Pro MoE on Ascend NPUs, a sparse model based on MoGE with 72 billion total parameters, 16 billion of which are activated for each token. The configuration of Pangu Pro MoE is optimized for Ascend 300I Duo and 800I A2 through extensive system simulation studies. Our experiments indicate that MoGE indeed leads to better expert load balancing and more efficient execution for both model training and inference on Ascend NPUs. The inference performance of Pangu Pro MoE achieves 1148 tokens/s per card and can be further improved to 1528 tokens/s per card by speculative acceleration, outperforming comparable 32B and 72B Dense models. Furthermore, we achieve an excellent cost-to-performance ratio for model inference on Ascend 300I Duo. Our studies show that Ascend NPUs are capable of training Pangu Pro MoE with massive parallelization to make it a leading model within the sub-100B total parameter class, outperforming prominent open-source models like GLM-Z1-32B and Qwen3-32B.
Pangu Light: Weight Re-Initialization for Pruning and Accelerating LLMs
May 26, 2025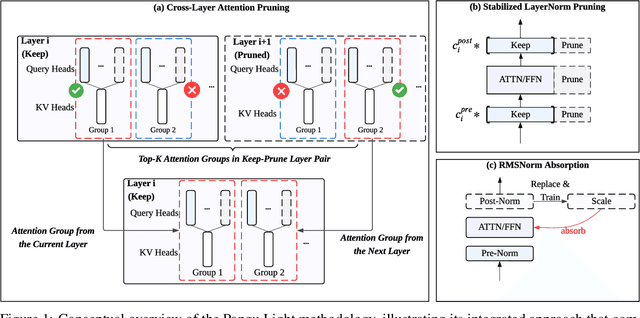

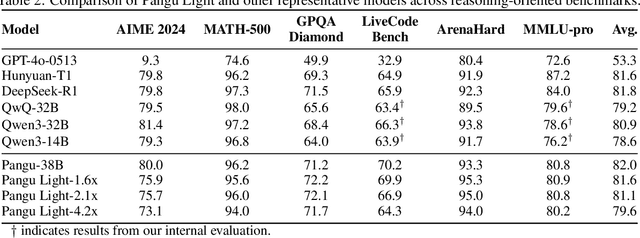
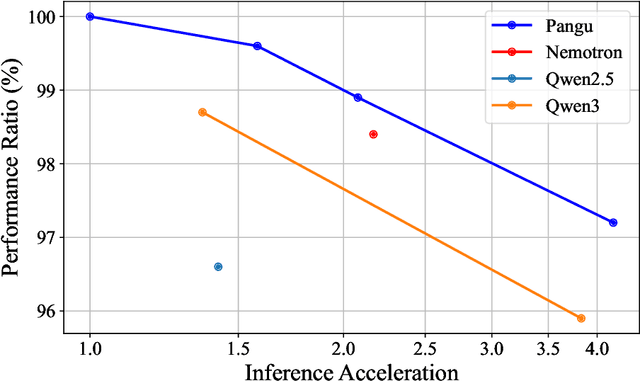
Large Language Models (LLMs) deliver state-of-the-art capabilities across numerous tasks, but their immense size and inference costs pose significant computational challenges for practical deployment. While structured pruning offers a promising avenue for model compression, existing methods often struggle with the detrimental effects of aggressive, simultaneous width and depth reductions, leading to substantial performance degradation. This paper argues that a critical, often overlooked, aspect in making such aggressive joint pruning viable is the strategic re-initialization and adjustment of remaining weights to improve the model post-pruning training accuracies. We introduce Pangu Light, a framework for LLM acceleration centered around structured pruning coupled with novel weight re-initialization techniques designed to address this ``missing piece''. Our framework systematically targets multiple axes, including model width, depth, attention heads, and RMSNorm, with its effectiveness rooted in novel re-initialization methods like Cross-Layer Attention Pruning (CLAP) and Stabilized LayerNorm Pruning (SLNP) that mitigate performance drops by providing the network a better training starting point. Further enhancing efficiency, Pangu Light incorporates specialized optimizations such as absorbing Post-RMSNorm computations and tailors its strategies to Ascend NPU characteristics. The Pangu Light models consistently exhibit a superior accuracy-efficiency trade-off, outperforming prominent baseline pruning methods like Nemotron and established LLMs like Qwen3 series. For instance, on Ascend NPUs, Pangu Light-32B's 81.6 average score and 2585 tokens/s throughput exceed Qwen3-32B's 80.9 average score and 2225 tokens/s.
Structure-Aligned Protein Language Model
May 22, 2025Protein language models (pLMs) pre-trained on vast protein sequence databases excel at various downstream tasks but lack the structural knowledge essential for many biological applications. To address this, we integrate structural insights from pre-trained protein graph neural networks (pGNNs) into pLMs through a latent-level contrastive learning task. This task aligns residue representations from pLMs with those from pGNNs across multiple proteins, enriching pLMs with inter-protein structural knowledge. Additionally, we incorporate a physical-level task that infuses intra-protein structural knowledge by optimizing pLMs to predict structural tokens. The proposed dual-task framework effectively incorporates both inter-protein and intra-protein structural knowledge into pLMs. Given the variability in the quality of protein structures in PDB, we further introduce a residue loss selection module, which uses a small model trained on high-quality structures to select reliable yet challenging residue losses for the pLM to learn. Applying our structure alignment method to the state-of-the-art ESM2 and AMPLIFY results in notable performance gains across a wide range of tasks, including a 12.7% increase in ESM2 contact prediction. The data, code, and resulting SaESM2 and SaAMPLIFY models will be released on Hugging Face.
Pangu Ultra MoE: How to Train Your Big MoE on Ascend NPUs
May 07, 2025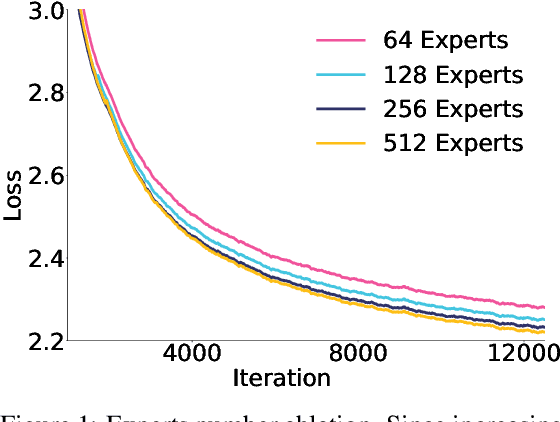

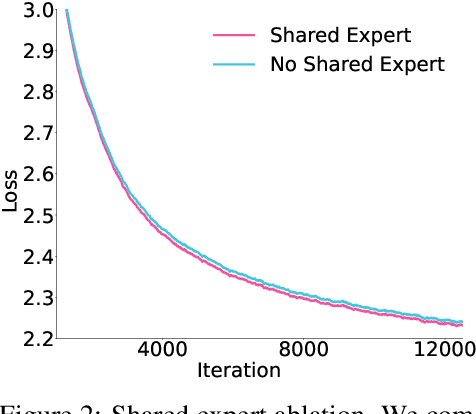

Sparse large language models (LLMs) with Mixture of Experts (MoE) and close to a trillion parameters are dominating the realm of most capable language models. However, the massive model scale poses significant challenges for the underlying software and hardware systems. In this paper, we aim to uncover a recipe to harness such scale on Ascend NPUs. The key goals are better usage of the computing resources under the dynamic sparse model structures and materializing the expected performance gain on the actual hardware. To select model configurations suitable for Ascend NPUs without repeatedly running the expensive experiments, we leverage simulation to compare the trade-off of various model hyperparameters. This study led to Pangu Ultra MoE, a sparse LLM with 718 billion parameters, and we conducted experiments on the model to verify the simulation results. On the system side, we dig into Expert Parallelism to optimize the communication between NPU devices to reduce the synchronization overhead. We also optimize the memory efficiency within the devices to further reduce the parameter and activation management overhead. In the end, we achieve an MFU of 30.0% when training Pangu Ultra MoE, with performance comparable to that of DeepSeek R1, on 6K Ascend NPUs, and demonstrate that the Ascend system is capable of harnessing all the training stages of the state-of-the-art language models. Extensive experiments indicate that our recipe can lead to efficient training of large-scale sparse language models with MoE. We also study the behaviors of such models for future reference.
Pangu Ultra: Pushing the Limits of Dense Large Language Models on Ascend NPUs
Apr 10, 2025



We present Pangu Ultra, a Large Language Model (LLM) with 135 billion parameters and dense Transformer modules trained on Ascend Neural Processing Units (NPUs). Although the field of LLM has been witnessing unprecedented advances in pushing the scale and capability of LLM in recent years, training such a large-scale model still involves significant optimization and system challenges. To stabilize the training process, we propose depth-scaled sandwich normalization, which effectively eliminates loss spikes during the training process of deep models. We pre-train our model on 13.2 trillion diverse and high-quality tokens and further enhance its reasoning capabilities during post-training. To perform such large-scale training efficiently, we utilize 8,192 Ascend NPUs with a series of system optimizations. Evaluations on multiple diverse benchmarks indicate that Pangu Ultra significantly advances the state-of-the-art capabilities of dense LLMs such as Llama 405B and Mistral Large 2, and even achieves competitive results with DeepSeek-R1, whose sparse model structure contains much more parameters. Our exploration demonstrates that Ascend NPUs are capable of efficiently and effectively training dense models with more than 100 billion parameters. Our model and system will be available for our commercial customers.
TransDiffSBDD: Causality-Aware Multi-Modal Structure-Based Drug Design
Mar 26, 2025Structure-based drug design (SBDD) is a critical task in drug discovery, requiring the generation of molecular information across two distinct modalities: discrete molecular graphs and continuous 3D coordinates. However, existing SBDD methods often overlook two key challenges: (1) the multi-modal nature of this task and (2) the causal relationship between these modalities, limiting their plausibility and performance. To address both challenges, we propose TransDiffSBDD, an integrated framework combining autoregressive transformers and diffusion models for SBDD. Specifically, the autoregressive transformer models discrete molecular information, while the diffusion model samples continuous distributions, effectively resolving the first challenge. To address the second challenge, we design a hybrid-modal sequence for protein-ligand complexes that explicitly respects the causality between modalities. Experiments on the CrossDocked2020 benchmark demonstrate that TransDiffSBDD outperforms existing baselines.
Offline Model-Based Optimization: Comprehensive Review
Mar 21, 2025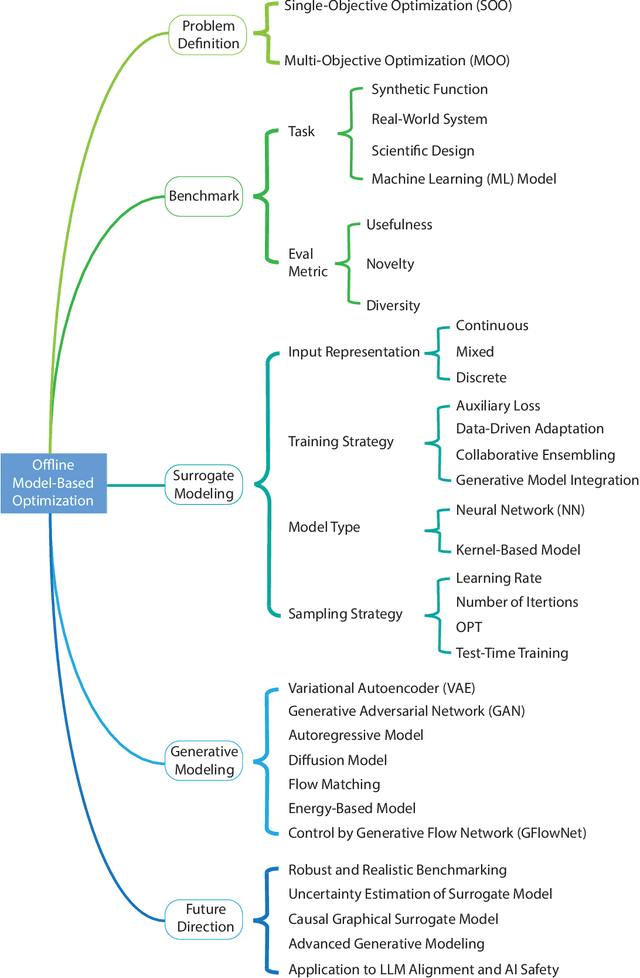
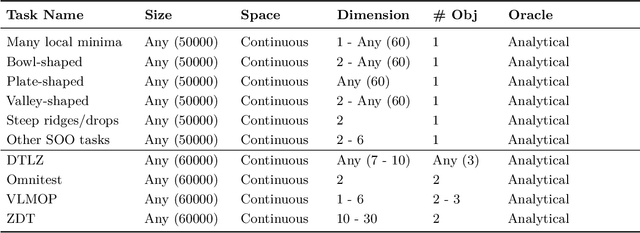
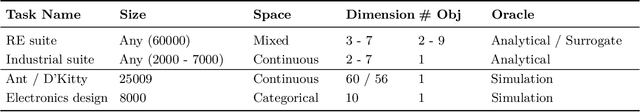
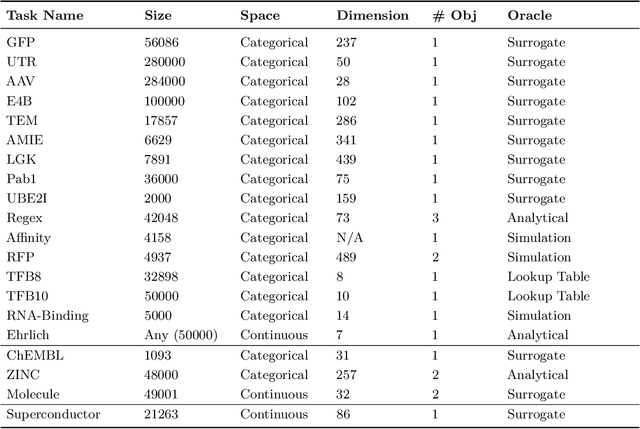
Offline optimization is a fundamental challenge in science and engineering, where the goal is to optimize black-box functions using only offline datasets. This setting is particularly relevant when querying the objective function is prohibitively expensive or infeasible, with applications spanning protein engineering, material discovery, neural architecture search, and beyond. The main difficulty lies in accurately estimating the objective landscape beyond the available data, where extrapolations are fraught with significant epistemic uncertainty. This uncertainty can lead to objective hacking(reward hacking), exploiting model inaccuracies in unseen regions, or other spurious optimizations that yield misleadingly high performance estimates outside the training distribution. Recent advances in model-based optimization(MBO) have harnessed the generalization capabilities of deep neural networks to develop offline-specific surrogate and generative models. Trained with carefully designed strategies, these models are more robust against out-of-distribution issues, facilitating the discovery of improved designs. Despite its growing impact in accelerating scientific discovery, the field lacks a comprehensive review. To bridge this gap, we present the first thorough review of offline MBO. We begin by formalizing the problem for both single-objective and multi-objective settings and by reviewing recent benchmarks and evaluation metrics. We then categorize existing approaches into two key areas: surrogate modeling, which emphasizes accurate function approximation in out-of-distribution regions, and generative modeling, which explores high-dimensional design spaces to identify high-performing designs. Finally, we examine the key challenges and propose promising directions for advancement in this rapidly evolving field including safe control of superintelligent systems.
Optimistic Interior Point Methods for Sequential Hypothesis Testing by Betting
Feb 11, 2025The technique of "testing by betting" frames nonparametric sequential hypothesis testing as a multiple-round game, where a player bets on future observations that arrive in a streaming fashion, accumulates wealth that quantifies evidence against the null hypothesis, and rejects the null once the wealth exceeds a specified threshold while controlling the false positive error. Designing an online learning algorithm that achieves a small regret in the game can help rapidly accumulate the bettor's wealth, which in turn can shorten the time to reject the null hypothesis under the alternative $H_1$. However, many of the existing works employ the Online Newton Step (ONS) to update within a halved decision space to avoid a gradient explosion issue, which is potentially conservative for rapid wealth accumulation. In this paper, we introduce a novel strategy utilizing interior-point methods in optimization that allows updates across the entire interior of the decision space without the risk of gradient explosion. Our approach not only maintains strong statistical guarantees but also facilitates faster null hypothesis rejection in critical scenarios, overcoming the limitations of existing approaches.
HODDI: A Dataset of High-Order Drug-Drug Interactions for Computational Pharmacovigilance
Feb 10, 2025
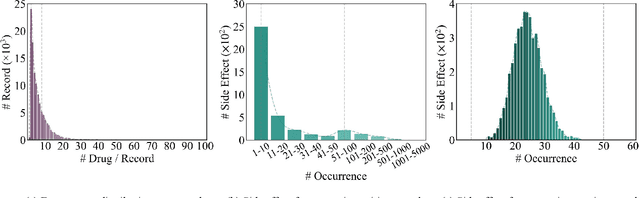
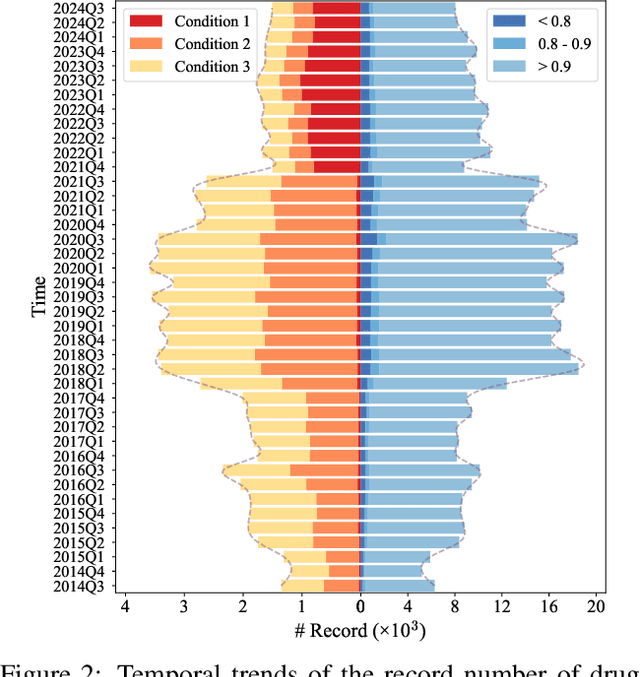
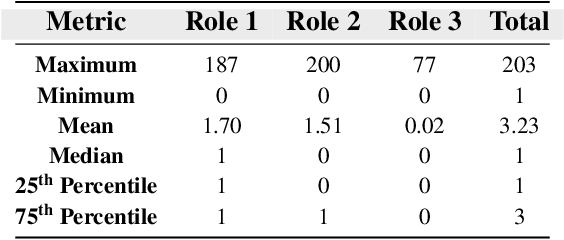
Drug-side effect research is vital for understanding adverse reactions arising in complex multi-drug therapies. However, the scarcity of higher-order datasets that capture the combinatorial effects of multiple drugs severely limits progress in this field. Existing resources such as TWOSIDES primarily focus on pairwise interactions. To fill this critical gap, we introduce HODDI, the first Higher-Order Drug-Drug Interaction Dataset, constructed from U.S. Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) records spanning the past decade, to advance computational pharmacovigilance. HODDI contains 109,744 records involving 2,506 unique drugs and 4,569 unique side effects, specifically curated to capture multi-drug interactions and their collective impact on adverse effects. Comprehensive statistical analyses demonstrate HODDI's extensive coverage and robust analytical metrics, making it a valuable resource for studying higher-order drug relationships. Evaluating HODDI with multiple models, we found that simple Multi-Layer Perceptron (MLP) can outperform graph models, while hypergraph models demonstrate superior performance in capturing complex multi-drug interactions, further validating HODDI's effectiveness. Our findings highlight the inherent value of higher-order information in drug-side effect prediction and position HODDI as a benchmark dataset for advancing research in pharmacovigilance, drug safety, and personalized medicine. The dataset and codes are available at https://github.com/TIML-Group/HODDI.