 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMosaicDoc: A Large-Scale Bilingual Benchmark for Visually Rich Document Understanding
Nov 13, 2025
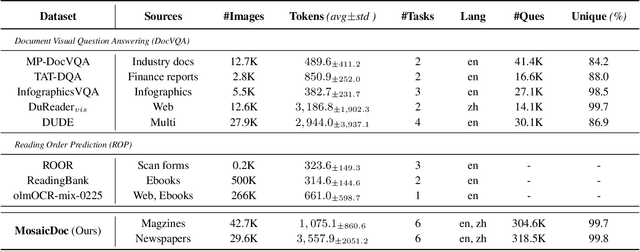
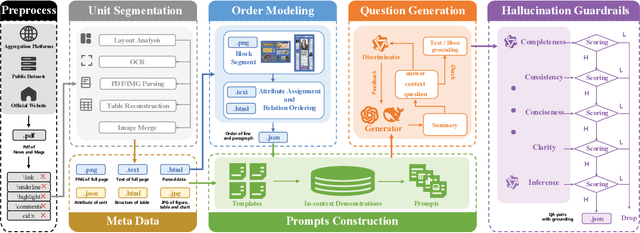
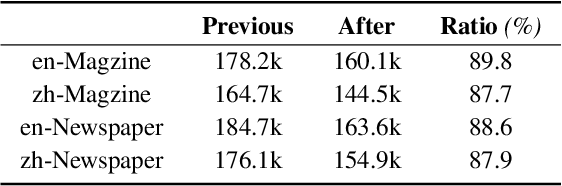
Despite the rapid progress of Vision-Language Models (VLMs), their capabilities are inadequately assessed by existing benchmarks, which are predominantly English-centric, feature simplistic layouts, and support limited tasks. Consequently, they fail to evaluate model performance for Visually Rich Document Understanding (VRDU), a critical challenge involving complex layouts and dense text. To address this, we introduce DocWeaver, a novel multi-agent pipeline that leverages Large Language Models to automatically generate a new benchmark. The result is MosaicDoc, a large-scale, bilingual (Chinese and English) resource designed to push the boundaries of VRDU. Sourced from newspapers and magazines, MosaicDoc features diverse and complex layouts (including multi-column and non-Manhattan), rich stylistic variety from 196 publishers, and comprehensive multi-task annotations (OCR, VQA, reading order, and localization). With 72K images and over 600K QA pairs, MosaicDoc serves as a definitive benchmark for the field. Our extensive evaluation of state-of-the-art models on this benchmark reveals their current limitations in handling real-world document complexity and charts a clear path for future research.
Precise Antigen-Antibody Structure Predictions Enhance Antibody Development with HelixFold-Multimer
Dec 13, 2024



The accurate prediction of antigen-antibody structures is essential for advancing immunology and therapeutic development, as it helps elucidate molecular interactions that underlie immune responses. Despite recent progress with deep learning models like AlphaFold and RoseTTAFold, accurately modeling antigen-antibody complexes remains a challenge due to their unique evolutionary characteristics. HelixFold-Multimer, a specialized model developed for this purpose, builds on the framework of AlphaFold-Multimer and demonstrates improved precision for antigen-antibody structures. HelixFold-Multimer not only surpasses other models in accuracy but also provides essential insights into antibody development, enabling more precise identification of binding sites, improved interaction prediction, and enhanced design of therapeutic antibodies. These advances underscore HelixFold-Multimer's potential in supporting antibody research and therapeutic innovation.
Technical Report of HelixFold3 for Biomolecular Structure Prediction
Aug 30, 2024



The AlphaFold series has transformed protein structure prediction with remarkable accuracy, often matching experimental methods. AlphaFold2, AlphaFold-Multimer, and the latest AlphaFold3 represent significant strides in predicting single protein chains, protein complexes, and biomolecular structures. While AlphaFold2 and AlphaFold-Multimer are open-sourced, facilitating rapid and reliable predictions, AlphaFold3 remains partially accessible through a limited online server and has not been open-sourced, restricting further development. To address these challenges, the PaddleHelix team is developing HelixFold3, aiming to replicate AlphaFold3's capabilities. Using insights from previous models and extensive datasets, HelixFold3 achieves an accuracy comparable to AlphaFold3 in predicting the structures of conventional ligands, nucleic acids, and proteins. The initial release of HelixFold3 is available as open source on GitHub for academic research, promising to advance biomolecular research and accelerate discoveries. We also provide online service at PaddleHelix website at https://paddlehelix.baidu.com/app/all/helixfold3/forecast.
Unifying Sequences, Structures, and Descriptions for Any-to-Any Protein Generation with the Large Multimodal Model HelixProtX
Jul 12, 2024Proteins are fundamental components of biological systems and can be represented through various modalities, including sequences, structures, and textual descriptions. Despite the advances in deep learning and scientific large language models (LLMs) for protein research, current methodologies predominantly focus on limited specialized tasks -- often predicting one protein modality from another. These approaches restrict the understanding and generation of multimodal protein data. In contrast, large multimodal models have demonstrated potential capabilities in generating any-to-any content like text, images, and videos, thus enriching user interactions across various domains. Integrating these multimodal model technologies into protein research offers significant promise by potentially transforming how proteins are studied. To this end, we introduce HelixProtX, a system built upon the large multimodal model, aiming to offer a comprehensive solution to protein research by supporting any-to-any protein modality generation. Unlike existing methods, it allows for the transformation of any input protein modality into any desired protein modality. The experimental results affirm the advanced capabilities of HelixProtX, not only in generating functional descriptions from amino acid sequences but also in executing critical tasks such as designing protein sequences and structures from textual descriptions. Preliminary findings indicate that HelixProtX consistently achieves superior accuracy across a range of protein-related tasks, outperforming existing state-of-the-art models. By integrating multimodal large models into protein research, HelixProtX opens new avenues for understanding protein biology, thereby promising to accelerate scientific discovery.
HelixFold-Multimer: Elevating Protein Complex Structure Prediction to New Heights
Apr 16, 2024



While monomer protein structure prediction tools boast impressive accuracy, the prediction of protein complex structures remains a daunting challenge in the field. This challenge is particularly pronounced in scenarios involving complexes with protein chains from different species, such as antigen-antibody interactions, where accuracy often falls short. Limited by the accuracy of complex prediction, tasks based on precise protein-protein interaction analysis also face obstacles. In this report, we highlight the ongoing advancements of our protein complex structure prediction model, HelixFold-Multimer, underscoring its enhanced performance. HelixFold-Multimer provides precise predictions for diverse protein complex structures, especially in therapeutic protein interactions. Notably, HelixFold-Multimer achieves remarkable success in antigen-antibody and peptide-protein structure prediction, surpassing AlphaFold-Multimer by several folds. HelixFold-Multimer is now available for public use on the PaddleHelix platform, offering both a general version and an antigen-antibody version. Researchers can conveniently access and utilize this service for their development needs.
GridFormer: Towards Accurate Table Structure Recognition via Grid Prediction
Sep 26, 2023



All tables can be represented as grids. Based on this observation, we propose GridFormer, a novel approach for interpreting unconstrained table structures by predicting the vertex and edge of a grid. First, we propose a flexible table representation in the form of an MXN grid. In this representation, the vertexes and edges of the grid store the localization and adjacency information of the table. Then, we introduce a DETR-style table structure recognizer to efficiently predict this multi-objective information of the grid in a single shot. Specifically, given a set of learned row and column queries, the recognizer directly outputs the vertexes and edges information of the corresponding rows and columns. Extensive experiments on five challenging benchmarks which include wired, wireless, multi-merge-cell, oriented, and distorted tables demonstrate the competitive performance of our model over other methods.
Online Map Vectorization for Autonomous Driving: A Rasterization Perspective
Jun 18, 2023



Vectorized high-definition (HD) map is essential for autonomous driving, providing detailed and precise environmental information for advanced perception and planning. However, current map vectorization methods often exhibit deviations, and the existing evaluation metric for map vectorization lacks sufficient sensitivity to detect these deviations. To address these limitations, we propose integrating the philosophy of rasterization into map vectorization. Specifically, we introduce a new rasterization-based evaluation metric, which has superior sensitivity and is better suited to real-world autonomous driving scenarios. Furthermore, we propose MapVR (Map Vectorization via Rasterization), a novel framework that applies differentiable rasterization to vectorized outputs and then performs precise and geometry-aware supervision on rasterized HD maps. Notably, MapVR designs tailored rasterization strategies for various geometric shapes, enabling effective adaptation to a wide range of map elements. Experiments show that incorporating rasterization into map vectorization greatly enhances performance with no extra computational cost during inference, leading to more accurate map perception and ultimately promoting safer autonomous driving.
HelixFold: An Efficient Implementation of AlphaFold2 using PaddlePaddle
Jul 13, 2022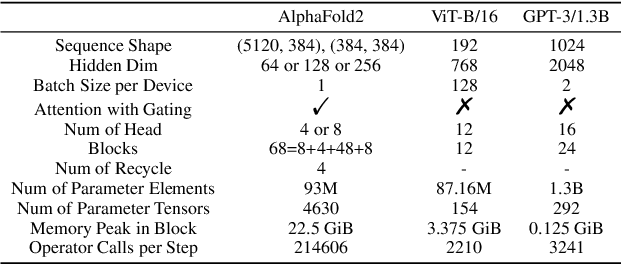
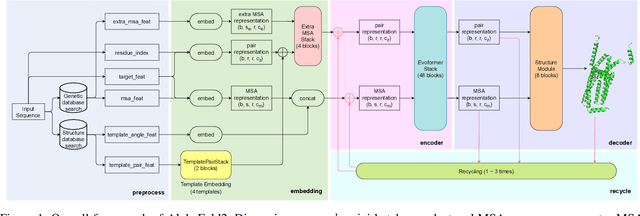
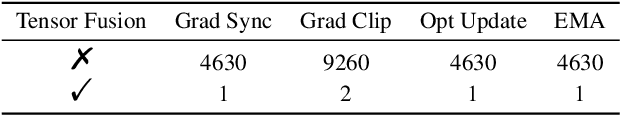
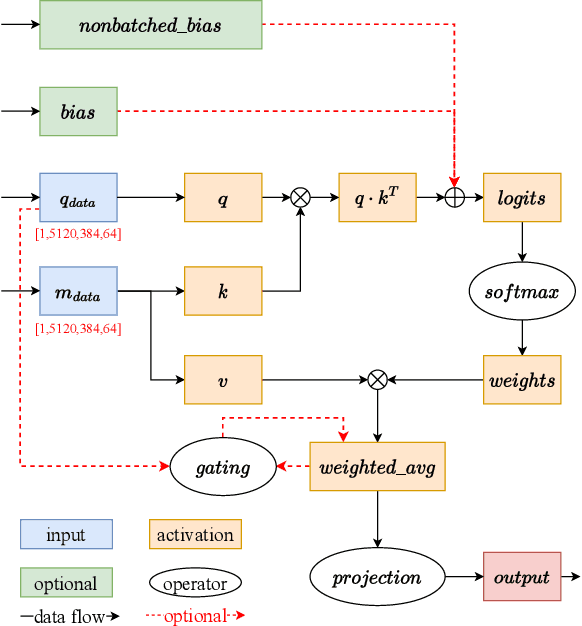
Accurate protein structure prediction can significantly accelerate the development of life science. The accuracy of AlphaFold2, a frontier end-to-end structure prediction system, is already close to that of the experimental determination techniques. Due to the complex model architecture and large memory consumption, it requires lots of computational resources and time to implement the training and inference of AlphaFold2 from scratch. The cost of running the original AlphaFold2 is expensive for most individuals and institutions. Therefore, reducing this cost could accelerate the development of life science. We implement AlphaFold2 using PaddlePaddle, namely HelixFold, to improve training and inference speed and reduce memory consumption. The performance is improved by operator fusion, tensor fusion, and hybrid parallelism computation, while the memory is optimized through Recompute, BFloat16, and memory read/write in-place. Compared with the original AlphaFold2 (implemented with Jax) and OpenFold (implemented with PyTorch), HelixFold needs only 7.5 days to complete the full end-to-end training and only 5.3 days when using hybrid parallelism, while both AlphaFold2 and OpenFold take about 11 days. HelixFold saves 1x training time. We verified that HelixFold's accuracy could be on par with AlphaFold2 on the CASP14 and CAMEO datasets. HelixFold's code is available on GitHub for free download: https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold, and we also provide stable web services on https://paddlehelix.baidu.com/app/drug/protein/forecast.
Bi-VLDoc: Bidirectional Vision-Language Modeling for Visually-Rich Document Understanding
Jun 27, 2022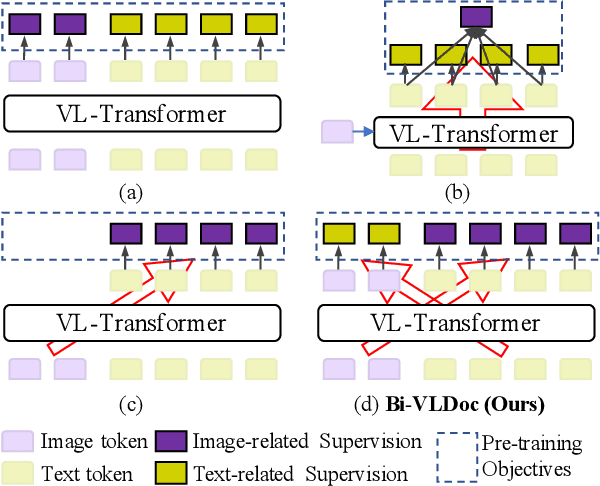
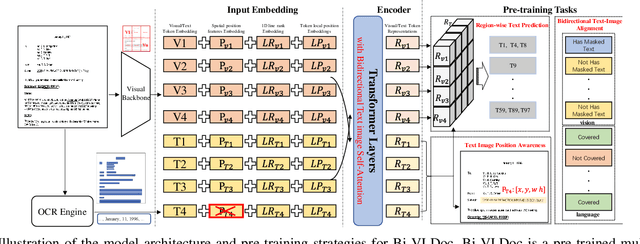
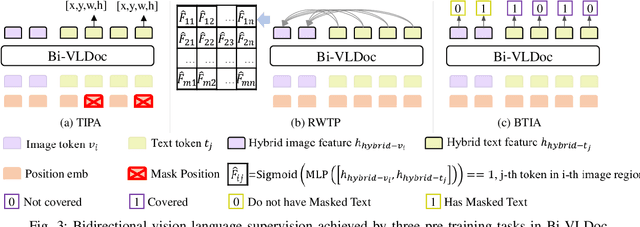
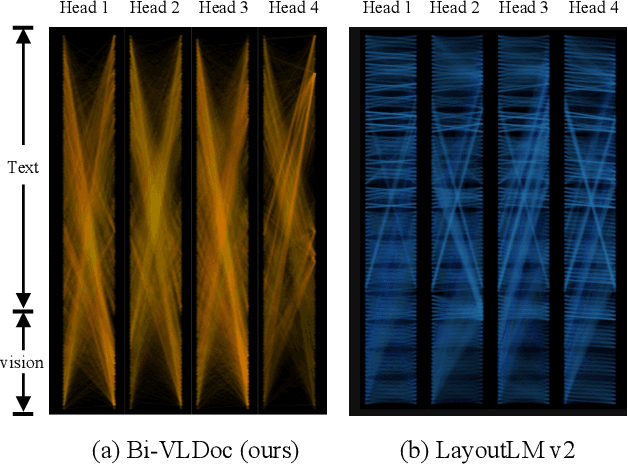
Multi-modal document pre-trained models have proven to be very effective in a variety of visually-rich document understanding (VrDU) tasks. Though existing document pre-trained models have achieved excellent performance on standard benchmarks for VrDU, the way they model and exploit the interactions between vision and language on documents has hindered them from better generalization ability and higher accuracy. In this work, we investigate the problem of vision-language joint representation learning for VrDU mainly from the perspective of supervisory signals. Specifically, a pre-training paradigm called Bi-VLDoc is proposed, in which a bidirectional vision-language supervision strategy and a vision-language hybrid-attention mechanism are devised to fully explore and utilize the interactions between these two modalities, to learn stronger cross-modal document representations with richer semantics. Benefiting from the learned informative cross-modal document representations, Bi-VLDoc significantly advances the state-of-the-art performance on three widely-used document understanding benchmarks, including Form Understanding (from 85.14% to 93.44%), Receipt Information Extraction (from 96.01% to 97.84%), and Document Classification (from 96.08% to 97.12%). On Document Visual QA, Bi-VLDoc achieves the state-of-the-art performance compared to previous single model methods.
GIRAFFE HD: A High-Resolution 3D-aware Generative Model
Mar 28, 2022



3D-aware generative models have shown that the introduction of 3D information can lead to more controllable image generation. In particular, the current state-of-the-art model GIRAFFE can control each object's rotation, translation, scale, and scene camera pose without corresponding supervision. However, GIRAFFE only operates well when the image resolution is low. We propose GIRAFFE HD, a high-resolution 3D-aware generative model that inherits all of GIRAFFE's controllable features while generating high-quality, high-resolution images ($512^2$ resolution and above). The key idea is to leverage a style-based neural renderer, and to independently generate the foreground and background to force their disentanglement while imposing consistency constraints to stitch them together to composite a coherent final image. We demonstrate state-of-the-art 3D controllable high-resolution image generation on multiple natural image datasets.