 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnd-to-End Reverse Screening Identifies Protein Targets of Small Molecules Using HelixFold3
Jan 20, 2026Identifying protein targets for small molecules, or reverse screening, is essential for understanding drug action, guiding compound repurposing, predicting off-target effects, and elucidating the molecular mechanisms of bioactive compounds. Despite its critical role, reverse screening remains challenging because accurately capturing interactions between a small molecule and structurally diverse proteins is inherently complex, and conventional step-wise workflows often propagate errors across decoupled steps such as target structure modeling, pocket identification, docking, and scoring. Here, we present an end-to-end reverse screening strategy leveraging HelixFold3, a high-accuracy biomolecular structure prediction model akin to AlphaFold3, which simultaneously models the folding of proteins from a protein library and the docking of small-molecule ligands within a unified framework. We validate this approach on a diverse and representative set of approximately one hundred small molecules. Compared with conventional reverse docking, our method improves screening accuracy and demonstrates enhanced structural fidelity, binding-site precision, and target prioritization. By systematically linking small molecules to their protein targets, this framework establishes a scalable and straightforward platform for dissecting molecular mechanisms, exploring off-target interactions, and supporting rational drug discovery.
Enhancing Q-Value Updates in Deep Q-Learning via Successor-State Prediction
Nov 05, 2025



Deep Q-Networks (DQNs) estimate future returns by learning from transitions sampled from a replay buffer. However, the target updates in DQN often rely on next states generated by actions from past, potentially suboptimal, policy. As a result, these states may not provide informative learning signals, causing high variance into the update process. This issue is exacerbated when the sampled transitions are poorly aligned with the agent's current policy. To address this limitation, we propose the Successor-state Aggregation Deep Q-Network (SADQ), which explicitly models environment dynamics using a stochastic transition model. SADQ integrates successor-state distributions into the Q-value estimation process, enabling more stable and policy-aligned value updates. Additionally, it explores a more efficient action selection strategy with the modeled transition structure. We provide theoretical guarantees that SADQ maintains unbiased value estimates while reducing training variance. Our extensive empirical results across standard RL benchmarks and real-world vector-based control tasks demonstrate that SADQ consistently outperforms DQN variants in both stability and learning efficiency.
From Static to Dynamic: Enhancing Offline-to-Online Reinforcement Learning via Energy-Guided Diffusion Stratification
Nov 05, 2025Transitioning from offline to online reinforcement learning (RL) poses critical challenges due to distributional shifts between the fixed behavior policy in the offline dataset and the evolving policy during online learning. Although this issue is widely recognized, few methods attempt to explicitly assess or utilize the distributional structure of the offline data itself, leaving a research gap in adapting learning strategies to different types of samples. To address this challenge, we propose an innovative method, Energy-Guided Diffusion Stratification (StratDiff), which facilitates smoother transitions in offline-to-online RL. StratDiff deploys a diffusion model to learn prior knowledge from the offline dataset. It then refines this knowledge through energy-based functions to improve policy imitation and generate offline-like actions during online fine-tuning. The KL divergence between the generated action and the corresponding sampled action is computed for each sample and used to stratify the training batch into offline-like and online-like subsets. Offline-like samples are updated using offline objectives, while online-like samples follow online learning strategies. We demonstrate the effectiveness of StratDiff by integrating it with off-the-shelf methods Cal-QL and IQL. Extensive empirical evaluations on D4RL benchmarks show that StratDiff significantly outperforms existing methods, achieving enhanced adaptability and more stable performance across diverse RL settings.
Behavior-Adaptive Q-Learning: A Unifying Framework for Offline-to-Online RL
Nov 05, 2025Offline reinforcement learning (RL) enables training from fixed data without online interaction, but policies learned offline often struggle when deployed in dynamic environments due to distributional shift and unreliable value estimates on unseen state-action pairs. We introduce Behavior-Adaptive Q-Learning (BAQ), a framework designed to enable a smooth and reliable transition from offline to online RL. The key idea is to leverage an implicit behavioral model derived from offline data to provide a behavior-consistency signal during online fine-tuning. BAQ incorporates a dual-objective loss that (i) aligns the online policy toward the offline behavior when uncertainty is high, and (ii) gradually relaxes this constraint as more confident online experience is accumulated. This adaptive mechanism reduces error propagation from out-of-distribution estimates, stabilizes early online updates, and accelerates adaptation to new scenarios. Across standard benchmarks, BAQ consistently outperforms prior offline-to-online RL approaches, achieving faster recovery, improved robustness, and higher overall performance. Our results demonstrate that implicit behavior adaptation is a principled and practical solution for reliable real-world policy deployment.
HelixDesign-Antibody: A Scalable Production-Grade Platform for Antibody Design Built on HelixFold3
Jul 03, 2025Antibody engineering is essential for developing therapeutics and advancing biomedical research. Traditional discovery methods often rely on time-consuming and resource-intensive experimental screening. To enhance and streamline this process, we introduce a production-grade, high-throughput platform built on HelixFold3, HelixDesign-Antibody, which utilizes the high-accuracy structure prediction model, HelixFold3. The platform facilitates the large-scale generation of antibody candidate sequences and evaluates their interaction with antigens. Integrated high-performance computing (HPC) support enables high-throughput screening, addressing challenges such as fragmented toolchains and high computational demands. Validation on multiple antigens showcases the platform's ability to generate diverse and high-quality antibodies, confirming a scaling law where exploring larger sequence spaces increases the likelihood of identifying optimal binders. This platform provides a seamless, accessible solution for large-scale antibody design and is available via the antibody design page of PaddleHelix platform.
HelixDesign-Binder: A Scalable Production-Grade Platform for Binder Design Built on HelixFold3
May 28, 2025Protein binder design is central to therapeutics, diagnostics, and synthetic biology, yet practical deployment remains challenging due to fragmented workflows, high computational costs, and complex tool integration. We present HelixDesign-Binder, a production-grade, high-throughput platform built on HelixFold3 that automates the full binder design pipeline, from backbone generation and sequence design to structural evaluation and multi-dimensional scoring. By unifying these stages into a scalable and user-friendly system, HelixDesign-Binder enables efficient exploration of binder candidates with favorable structural, energetic, and physicochemical properties. The platform leverages Baidu Cloud's high-performance infrastructure to support large-scale design and incorporates advanced scoring metrics, including ipTM, predicted binding free energy, and interface hydrophobicity. Benchmarking across six protein targets demonstrates that HelixDesign-Binder reliably produces diverse and high-quality binders, some of which match or exceed validated designs in predicted binding affinity. HelixDesign-Binder is accessible via an interactive web interface in PaddleHelix platform, supporting both academic research and industrial applications in antibody and protein binder development.
Precise Antigen-Antibody Structure Predictions Enhance Antibody Development with HelixFold-Multimer
Dec 13, 2024



The accurate prediction of antigen-antibody structures is essential for advancing immunology and therapeutic development, as it helps elucidate molecular interactions that underlie immune responses. Despite recent progress with deep learning models like AlphaFold and RoseTTAFold, accurately modeling antigen-antibody complexes remains a challenge due to their unique evolutionary characteristics. HelixFold-Multimer, a specialized model developed for this purpose, builds on the framework of AlphaFold-Multimer and demonstrates improved precision for antigen-antibody structures. HelixFold-Multimer not only surpasses other models in accuracy but also provides essential insights into antibody development, enabling more precise identification of binding sites, improved interaction prediction, and enhanced design of therapeutic antibodies. These advances underscore HelixFold-Multimer's potential in supporting antibody research and therapeutic innovation.
Technical Report of HelixFold3 for Biomolecular Structure Prediction
Aug 30, 2024



The AlphaFold series has transformed protein structure prediction with remarkable accuracy, often matching experimental methods. AlphaFold2, AlphaFold-Multimer, and the latest AlphaFold3 represent significant strides in predicting single protein chains, protein complexes, and biomolecular structures. While AlphaFold2 and AlphaFold-Multimer are open-sourced, facilitating rapid and reliable predictions, AlphaFold3 remains partially accessible through a limited online server and has not been open-sourced, restricting further development. To address these challenges, the PaddleHelix team is developing HelixFold3, aiming to replicate AlphaFold3's capabilities. Using insights from previous models and extensive datasets, HelixFold3 achieves an accuracy comparable to AlphaFold3 in predicting the structures of conventional ligands, nucleic acids, and proteins. The initial release of HelixFold3 is available as open source on GitHub for academic research, promising to advance biomolecular research and accelerate discoveries. We also provide online service at PaddleHelix website at https://paddlehelix.baidu.com/app/all/helixfold3/forecast.
FedAR: Addressing Client Unavailability in Federated Learning with Local Update Approximation and Rectification
Jul 26, 2024

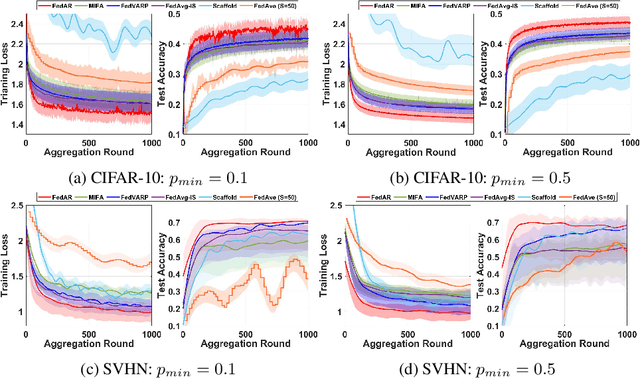

Federated learning (FL) enables clients to collaboratively train machine learning models under the coordination of a server in a privacy-preserving manner. One of the main challenges in FL is that the server may not receive local updates from each client in each round due to client resource limitations and intermittent network connectivity. The existence of unavailable clients severely deteriorates the overall FL performance. In this paper, we propose , a novel client update Approximation and Rectification algorithm for FL to address the client unavailability issue. FedAR can get all clients involved in the global model update to achieve a high-quality global model on the server, which also furnishes accurate predictions for each client. To this end, the server uses the latest update from each client as a surrogate for its current update. It then assigns a different weight to each client's surrogate update to derive the global model, in order to guarantee contributions from both available and unavailable clients. Our theoretical analysis proves that FedAR achieves optimal convergence rates on non-IID datasets for both convex and non-convex smooth loss functions. Extensive empirical studies show that FedAR comprehensively outperforms state-of-the-art FL baselines including FedAvg, MIFA, FedVARP and Scaffold in terms of the training loss, test accuracy, and bias mitigation. Moreover, FedAR also depicts impressive performance in the presence of a large number of clients with severe client unavailability.
Unifying Sequences, Structures, and Descriptions for Any-to-Any Protein Generation with the Large Multimodal Model HelixProtX
Jul 12, 2024Proteins are fundamental components of biological systems and can be represented through various modalities, including sequences, structures, and textual descriptions. Despite the advances in deep learning and scientific large language models (LLMs) for protein research, current methodologies predominantly focus on limited specialized tasks -- often predicting one protein modality from another. These approaches restrict the understanding and generation of multimodal protein data. In contrast, large multimodal models have demonstrated potential capabilities in generating any-to-any content like text, images, and videos, thus enriching user interactions across various domains. Integrating these multimodal model technologies into protein research offers significant promise by potentially transforming how proteins are studied. To this end, we introduce HelixProtX, a system built upon the large multimodal model, aiming to offer a comprehensive solution to protein research by supporting any-to-any protein modality generation. Unlike existing methods, it allows for the transformation of any input protein modality into any desired protein modality. The experimental results affirm the advanced capabilities of HelixProtX, not only in generating functional descriptions from amino acid sequences but also in executing critical tasks such as designing protein sequences and structures from textual descriptions. Preliminary findings indicate that HelixProtX consistently achieves superior accuracy across a range of protein-related tasks, outperforming existing state-of-the-art models. By integrating multimodal large models into protein research, HelixProtX opens new avenues for understanding protein biology, thereby promising to accelerate scientific discovery.