 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrecise Antigen-Antibody Structure Predictions Enhance Antibody Development with HelixFold-Multimer
Dec 13, 2024



The accurate prediction of antigen-antibody structures is essential for advancing immunology and therapeutic development, as it helps elucidate molecular interactions that underlie immune responses. Despite recent progress with deep learning models like AlphaFold and RoseTTAFold, accurately modeling antigen-antibody complexes remains a challenge due to their unique evolutionary characteristics. HelixFold-Multimer, a specialized model developed for this purpose, builds on the framework of AlphaFold-Multimer and demonstrates improved precision for antigen-antibody structures. HelixFold-Multimer not only surpasses other models in accuracy but also provides essential insights into antibody development, enabling more precise identification of binding sites, improved interaction prediction, and enhanced design of therapeutic antibodies. These advances underscore HelixFold-Multimer's potential in supporting antibody research and therapeutic innovation.
Technical Report of HelixFold3 for Biomolecular Structure Prediction
Aug 30, 2024



The AlphaFold series has transformed protein structure prediction with remarkable accuracy, often matching experimental methods. AlphaFold2, AlphaFold-Multimer, and the latest AlphaFold3 represent significant strides in predicting single protein chains, protein complexes, and biomolecular structures. While AlphaFold2 and AlphaFold-Multimer are open-sourced, facilitating rapid and reliable predictions, AlphaFold3 remains partially accessible through a limited online server and has not been open-sourced, restricting further development. To address these challenges, the PaddleHelix team is developing HelixFold3, aiming to replicate AlphaFold3's capabilities. Using insights from previous models and extensive datasets, HelixFold3 achieves an accuracy comparable to AlphaFold3 in predicting the structures of conventional ligands, nucleic acids, and proteins. The initial release of HelixFold3 is available as open source on GitHub for academic research, promising to advance biomolecular research and accelerate discoveries. We also provide online service at PaddleHelix website at https://paddlehelix.baidu.com/app/all/helixfold3/forecast.
HelixFold-Multimer: Elevating Protein Complex Structure Prediction to New Heights
Apr 16, 2024



While monomer protein structure prediction tools boast impressive accuracy, the prediction of protein complex structures remains a daunting challenge in the field. This challenge is particularly pronounced in scenarios involving complexes with protein chains from different species, such as antigen-antibody interactions, where accuracy often falls short. Limited by the accuracy of complex prediction, tasks based on precise protein-protein interaction analysis also face obstacles. In this report, we highlight the ongoing advancements of our protein complex structure prediction model, HelixFold-Multimer, underscoring its enhanced performance. HelixFold-Multimer provides precise predictions for diverse protein complex structures, especially in therapeutic protein interactions. Notably, HelixFold-Multimer achieves remarkable success in antigen-antibody and peptide-protein structure prediction, surpassing AlphaFold-Multimer by several folds. HelixFold-Multimer is now available for public use on the PaddleHelix platform, offering both a general version and an antigen-antibody version. Researchers can conveniently access and utilize this service for their development needs.
Pre-Training on Large-Scale Generated Docking Conformations with HelixDock to Unlock the Potential of Protein-ligand Structure Prediction Models
Oct 21, 2023Molecular docking, a pivotal computational tool for drug discovery, predicts the binding interactions between small molecules (ligands) and target proteins (receptors). Conventional physics-based docking tools, though widely used, face limitations in precision due to restricted conformational sampling and imprecise scoring functions. Recent endeavors have employed deep learning techniques to enhance docking accuracy, but their generalization remains a concern due to limited training data. Leveraging the success of extensive and diverse data in other domains, we introduce HelixDock, a novel approach for site-specific molecular docking. Hundreds of millions of binding poses are generated by traditional docking tools, encompassing diverse protein targets and small molecules. Our deep learning-based docking model, a SE(3)-equivariant network, is pre-trained with this large-scale dataset and then fine-tuned with a small number of precise receptor-ligand complex structures. Comparative analyses against physics-based and deep learning-based baseline methods highlight HelixDock's superiority, especially on challenging test sets. Our study elucidates the scaling laws of the pre-trained molecular docking models, showcasing consistent improvements with increased model parameters and pre-train data quantities. Harnessing the power of extensive and diverse generated data holds promise for advancing AI-driven drug discovery.
GEM-2: Next Generation Molecular Property Prediction Network with Many-body and Full-range Interaction Modeling
Aug 15, 2022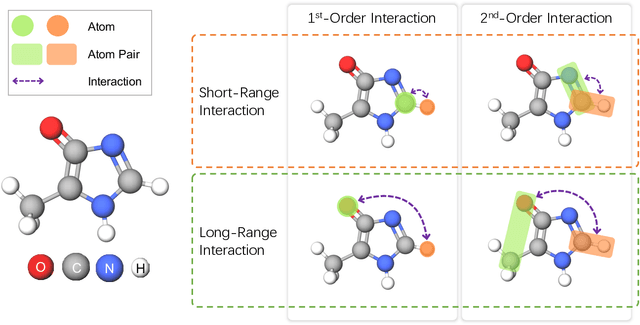
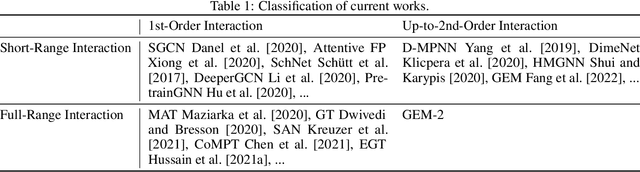
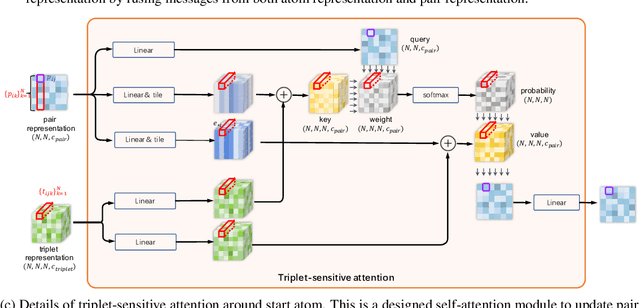
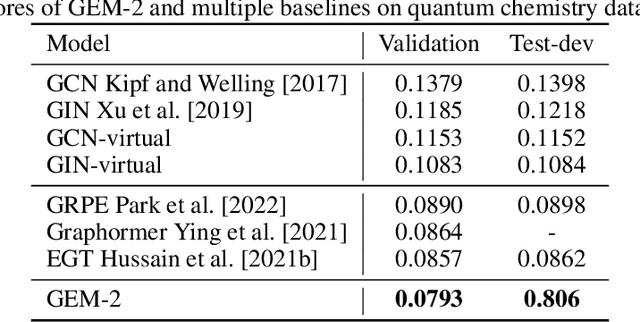
Molecular property prediction is a fundamental task in the drug and material industries. Physically, the properties of a molecule are determined by its own electronic structure, which can be exactly described by the Schr\"odinger equation. However, solving the Schr\"odinger equation for most molecules is extremely challenging due to long-range interactions in the behavior of a quantum many-body system. While deep learning methods have proven to be effective in molecular property prediction, we design a novel method, namely GEM-2, which comprehensively considers both the long-range and many-body interactions in molecules. GEM-2 consists of two interacted tracks: an atom-level track modeling both the local and global correlation between any two atoms, and a pair-level track modeling the correlation between all atom pairs, which embed information between any 3 or 4 atoms. Extensive experiments demonstrated the superiority of GEM-2 over multiple baseline methods in quantum chemistry and drug discovery tasks.
HelixFold-Single: MSA-free Protein Structure Prediction by Using Protein Language Model as an Alternative
Aug 09, 2022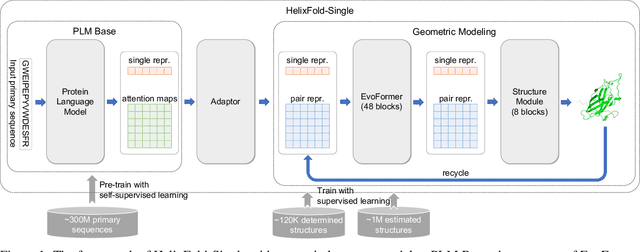


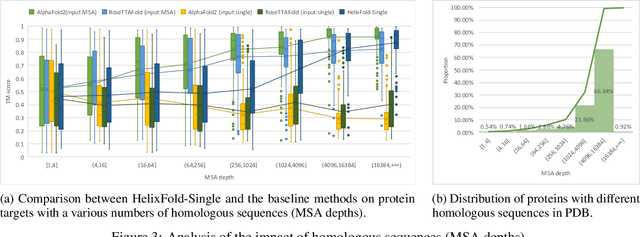
AI-based protein structure prediction pipelines, such as AlphaFold2, have achieved near-experimental accuracy. These advanced pipelines mainly rely on Multiple Sequence Alignments (MSAs) as inputs to learn the co-evolution information from the homologous sequences. Nonetheless, searching MSAs from protein databases is time-consuming, usually taking dozens of minutes. Consequently, we attempt to explore the limits of fast protein structure prediction by using only primary sequences of proteins. HelixFold-Single is proposed to combine a large-scale protein language model with the superior geometric learning capability of AlphaFold2. Our proposed method, HelixFold-Single, first pre-trains a large-scale protein language model (PLM) with thousands of millions of primary sequences utilizing the self-supervised learning paradigm, which will be used as an alternative to MSAs for learning the co-evolution information. Then, by combining the pre-trained PLM and the essential components of AlphaFold2, we obtain an end-to-end differentiable model to predict the 3D coordinates of atoms from only the primary sequence. HelixFold-Single is validated in datasets CASP14 and CAMEO, achieving competitive accuracy with the MSA-based methods on the targets with large homologous families. Furthermore, HelixFold-Single consumes much less time than the mainstream pipelines for protein structure prediction, demonstrating its potential in tasks requiring many predictions. The code of HelixFold-Single is available at https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold-single, and we also provide stable web services on https://paddlehelix.baidu.com/app/drug/protein-single/forecast.
HelixADMET: a robust and endpoint extensible ADMET system incorporating self-supervised knowledge transfer
May 17, 2022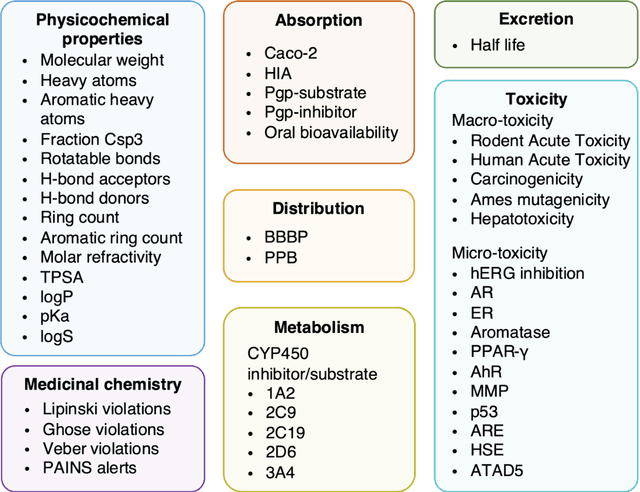
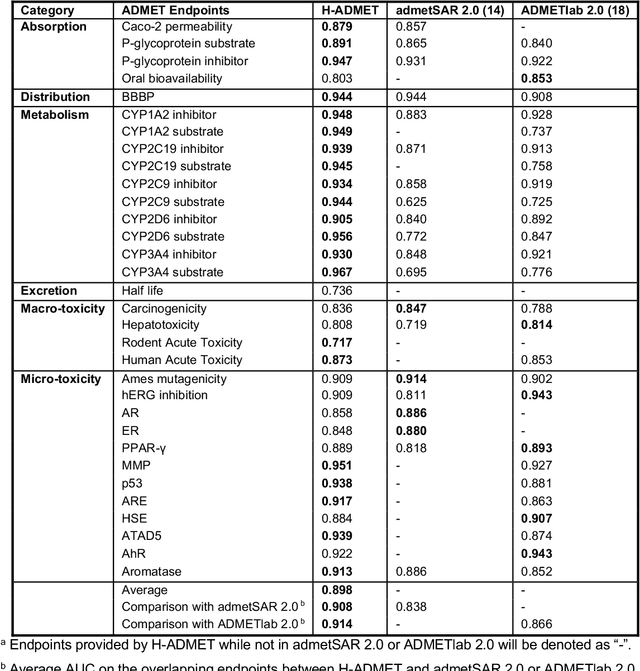
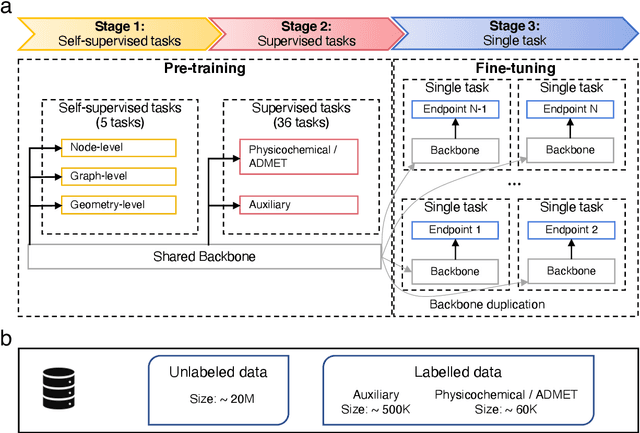
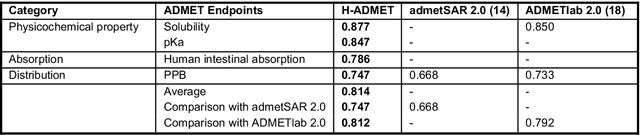
Accurate ADMET (an abbreviation for "absorption, distribution, metabolism, excretion, and toxicity") predictions can efficiently screen out undesirable drug candidates in the early stage of drug discovery. In recent years, multiple comprehensive ADMET systems that adopt advanced machine learning models have been developed, providing services to estimate multiple endpoints. However, those ADMET systems usually suffer from weak extrapolation ability. First, due to the lack of labelled data for each endpoint, typical machine learning models perform frail for the molecules with unobserved scaffolds. Second, most systems only provide fixed built-in endpoints and cannot be customised to satisfy various research requirements. To this end, we develop a robust and endpoint extensible ADMET system, HelixADMET (H-ADMET). H-ADMET incorporates the concept of self-supervised learning to produce a robust pre-trained model. The model is then fine-tuned with a multi-task and multi-stage framework to transfer knowledge between ADMET endpoints, auxiliary tasks, and self-supervised tasks. Our results demonstrate that H-ADMET achieves an overall improvement of 4%, compared with existing ADMET systems on comparable endpoints. Additionally, the pre-trained model provided by H-ADMET can be fine-tuned to generate new and customised ADMET endpoints, meeting various demands of drug research and development requirements.
ChemRL-GEM: Geometry Enhanced Molecular Representation Learning for Property Prediction
Jul 08, 2021
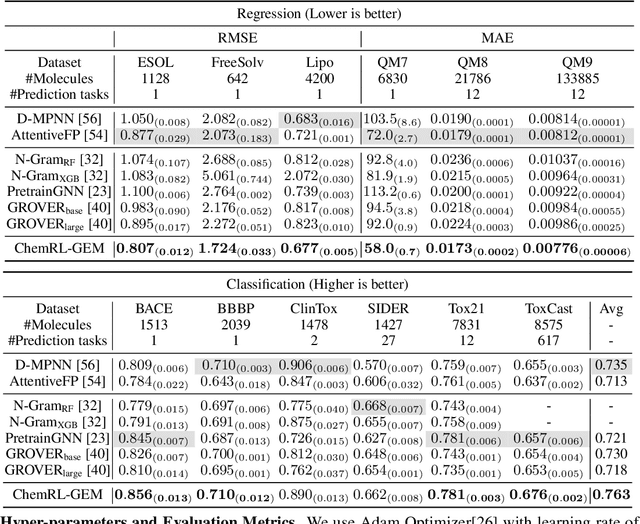
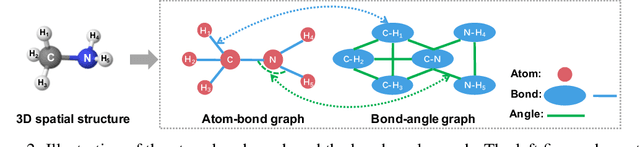
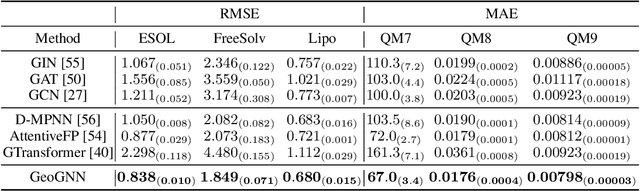
Effective molecular representation learning is of great importance to facilitate molecular property prediction, which is a fundamental task for the drug and material industry. Recent advances in graph neural networks (GNNs) have shown great promise in applying GNNs for molecular representation learning. Moreover, a few recent studies have also demonstrated successful applications of self-supervised learning methods to pre-train the GNNs to overcome the problem of insufficient labeled molecules. However, existing GNNs and pre-training strategies usually treat molecules as topological graph data without fully utilizing the molecular geometry information. Whereas, the three-dimensional (3D) spatial structure of a molecule, a.k.a molecular geometry, is one of the most critical factors for determining molecular physical, chemical, and biological properties. To this end, we propose a novel Geometry Enhanced Molecular representation learning method (GEM) for Chemical Representation Learning (ChemRL). At first, we design a geometry-based GNN architecture that simultaneously models atoms, bonds, and bond angles in a molecule. To be specific, we devised double graphs for a molecule: The first one encodes the atom-bond relations; The second one encodes bond-angle relations. Moreover, on top of the devised GNN architecture, we propose several novel geometry-level self-supervised learning strategies to learn spatial knowledge by utilizing the local and global molecular 3D structures. We compare ChemRL-GEM with various state-of-the-art (SOTA) baselines on different molecular benchmarks and exhibit that ChemRL-GEM can significantly outperform all baselines in both regression and classification tasks. For example, the experimental results show an overall improvement of 8.8% on average compared to SOTA baselines on the regression tasks, demonstrating the superiority of the proposed method.
LiteGEM: Lite Geometry Enhanced Molecular Representation Learning for Quantum Property Prediction
Jun 28, 2021

In this report, we (SuperHelix team) present our solution to KDD Cup 2021-PCQM4M-LSC, a large-scale quantum chemistry dataset on predicting HOMO-LUMO gap of molecules. Our solution, Lite Geometry Enhanced Molecular representation learning (LiteGEM) achieves a mean absolute error (MAE) of 0.1204 on the test set with the help of deep graph neural networks and various self-supervised learning tasks. The code of the framework can be found in https://github.com/PaddlePaddle/PaddleHelix/tree/dev/competition/kddcup2021-PCQM4M-LSC/.
MBCAL: A Simple and Efficient Reinforcement Learning Method for Recommendation Systems
Nov 06, 2019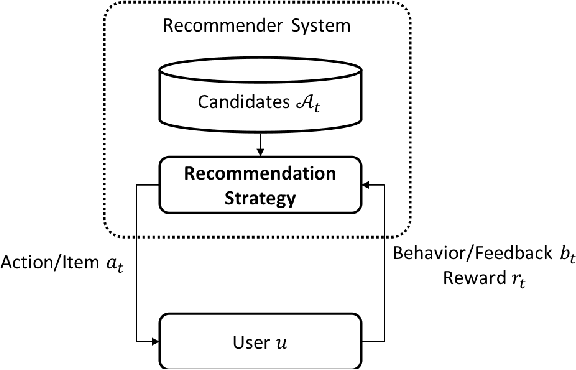
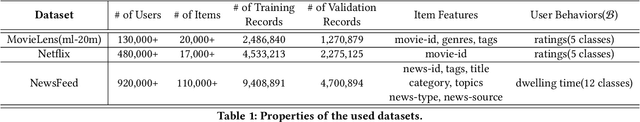
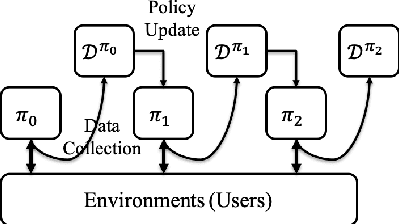
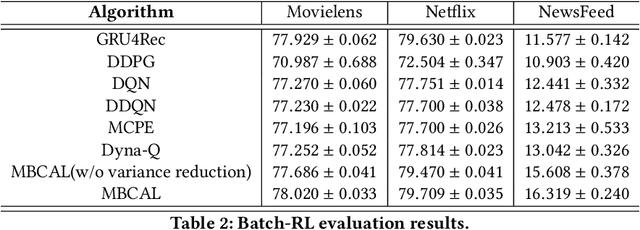
It has been widely regarded that only considering the immediate user feedback is not sufficient for modern industrial recommendation systems. Many previous works attempt to maximize the long term rewards with Reinforcement Learning(RL). However, model-free RL suffers from problems including significant variance in gradient, long convergence period, and requirement of sophisticated online infrastructures. While model-based RL provides a sample-efficient choice, the cost of planning in an online system is unacceptable. To achieve high sample efficiency in practical situations, we propose a novel model-based reinforcement learning method, namely the model-based counterfactual advantage learning(MBCAL). In the proposed method, a masking item is introduced in the environment model learning. With the masking item and the environment model, we introduce the counterfactual future advantage, which eliminates most of the noises in long term rewards. The proposed method selects through approximating the immediate reward and future advantage separately. It is easy to implement, yet it requires reasonable cost in both training and inference processes. In the experiments, we compare our methods with several baselines, including supervised learning, model-free RL, and other model-based RL methods in carefully designed experiments. Results show that our method transcends all the baselines in both sample efficiency and asymptotic performance.