 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePre-Training on Large-Scale Generated Docking Conformations with HelixDock to Unlock the Potential of Protein-ligand Structure Prediction Models
Oct 21, 2023Molecular docking, a pivotal computational tool for drug discovery, predicts the binding interactions between small molecules (ligands) and target proteins (receptors). Conventional physics-based docking tools, though widely used, face limitations in precision due to restricted conformational sampling and imprecise scoring functions. Recent endeavors have employed deep learning techniques to enhance docking accuracy, but their generalization remains a concern due to limited training data. Leveraging the success of extensive and diverse data in other domains, we introduce HelixDock, a novel approach for site-specific molecular docking. Hundreds of millions of binding poses are generated by traditional docking tools, encompassing diverse protein targets and small molecules. Our deep learning-based docking model, a SE(3)-equivariant network, is pre-trained with this large-scale dataset and then fine-tuned with a small number of precise receptor-ligand complex structures. Comparative analyses against physics-based and deep learning-based baseline methods highlight HelixDock's superiority, especially on challenging test sets. Our study elucidates the scaling laws of the pre-trained molecular docking models, showcasing consistent improvements with increased model parameters and pre-train data quantities. Harnessing the power of extensive and diverse generated data holds promise for advancing AI-driven drug discovery.
GEM-2: Next Generation Molecular Property Prediction Network with Many-body and Full-range Interaction Modeling
Aug 15, 2022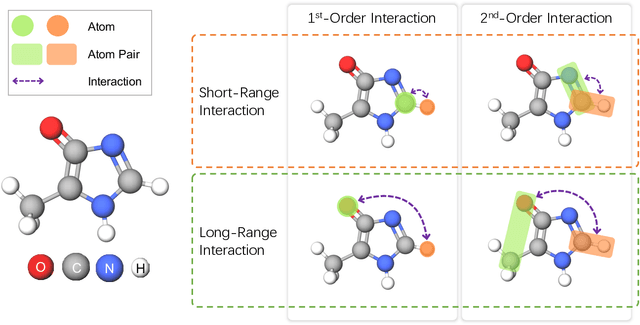
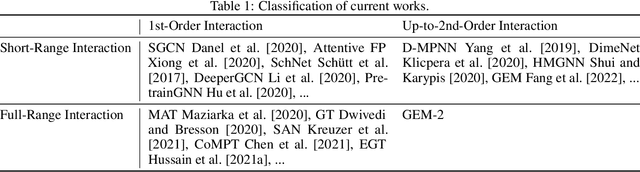
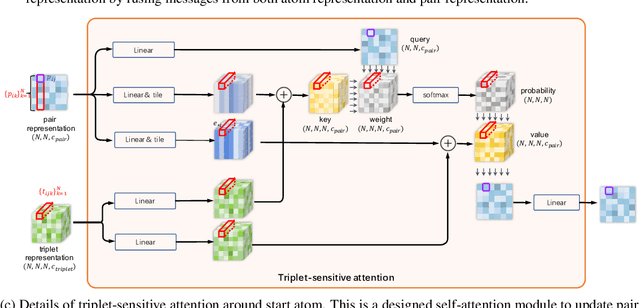
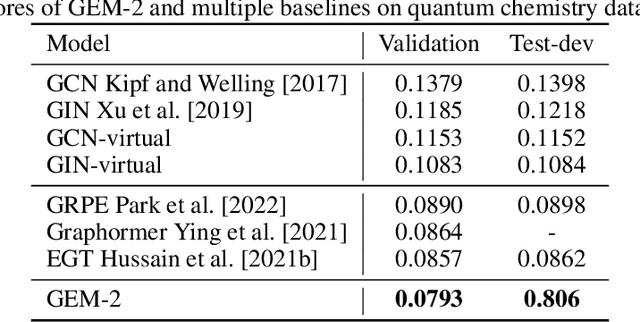
Molecular property prediction is a fundamental task in the drug and material industries. Physically, the properties of a molecule are determined by its own electronic structure, which can be exactly described by the Schr\"odinger equation. However, solving the Schr\"odinger equation for most molecules is extremely challenging due to long-range interactions in the behavior of a quantum many-body system. While deep learning methods have proven to be effective in molecular property prediction, we design a novel method, namely GEM-2, which comprehensively considers both the long-range and many-body interactions in molecules. GEM-2 consists of two interacted tracks: an atom-level track modeling both the local and global correlation between any two atoms, and a pair-level track modeling the correlation between all atom pairs, which embed information between any 3 or 4 atoms. Extensive experiments demonstrated the superiority of GEM-2 over multiple baseline methods in quantum chemistry and drug discovery tasks.
HelixADMET: a robust and endpoint extensible ADMET system incorporating self-supervised knowledge transfer
May 17, 2022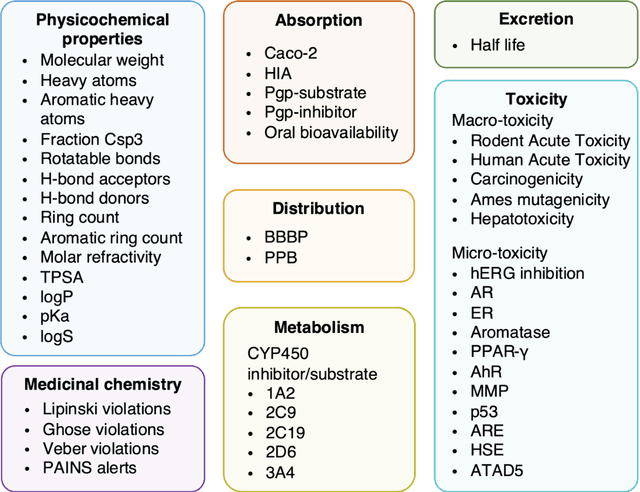
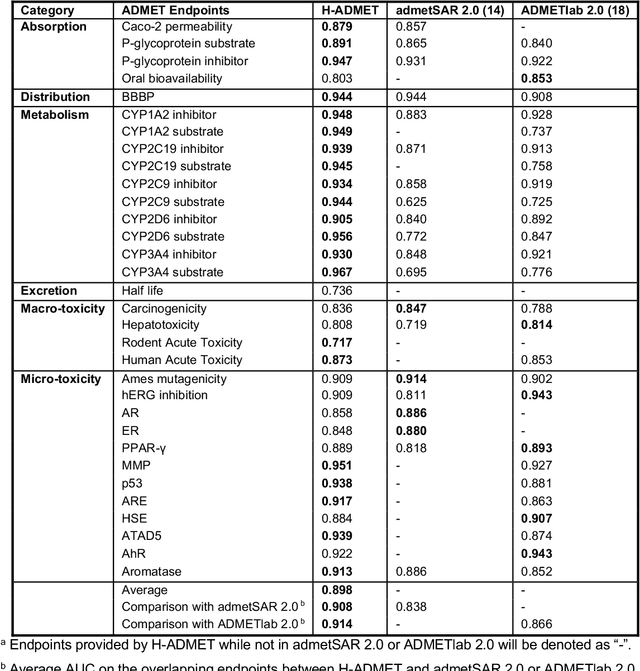
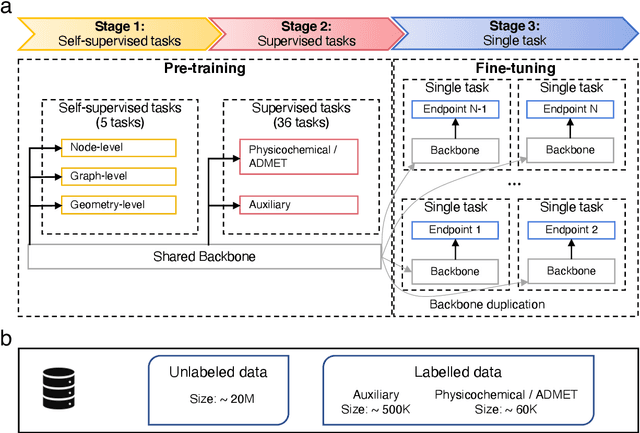
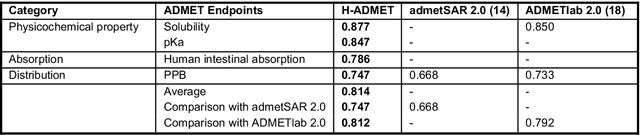
Accurate ADMET (an abbreviation for "absorption, distribution, metabolism, excretion, and toxicity") predictions can efficiently screen out undesirable drug candidates in the early stage of drug discovery. In recent years, multiple comprehensive ADMET systems that adopt advanced machine learning models have been developed, providing services to estimate multiple endpoints. However, those ADMET systems usually suffer from weak extrapolation ability. First, due to the lack of labelled data for each endpoint, typical machine learning models perform frail for the molecules with unobserved scaffolds. Second, most systems only provide fixed built-in endpoints and cannot be customised to satisfy various research requirements. To this end, we develop a robust and endpoint extensible ADMET system, HelixADMET (H-ADMET). H-ADMET incorporates the concept of self-supervised learning to produce a robust pre-trained model. The model is then fine-tuned with a multi-task and multi-stage framework to transfer knowledge between ADMET endpoints, auxiliary tasks, and self-supervised tasks. Our results demonstrate that H-ADMET achieves an overall improvement of 4%, compared with existing ADMET systems on comparable endpoints. Additionally, the pre-trained model provided by H-ADMET can be fine-tuned to generate new and customised ADMET endpoints, meeting various demands of drug research and development requirements.
ChemRL-GEM: Geometry Enhanced Molecular Representation Learning for Property Prediction
Jul 08, 2021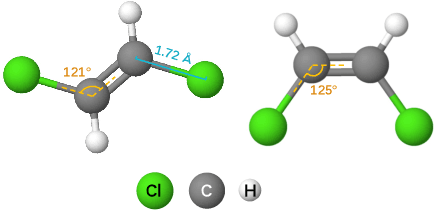
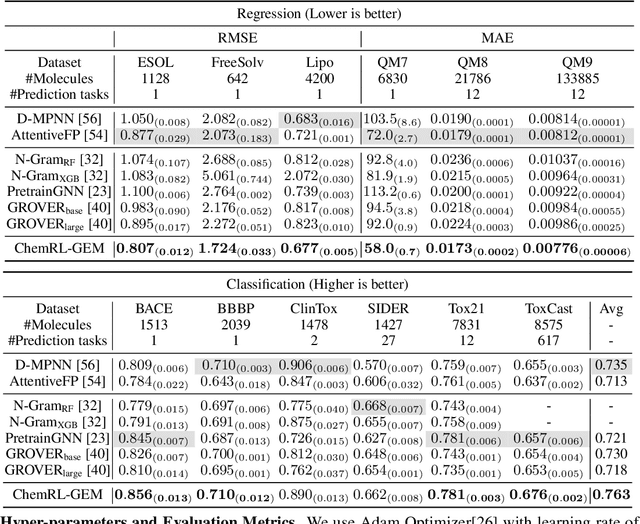
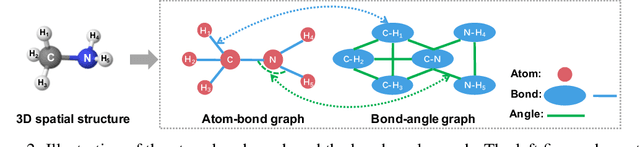
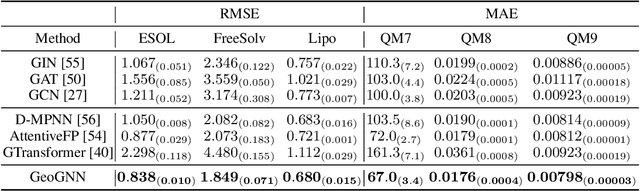
Effective molecular representation learning is of great importance to facilitate molecular property prediction, which is a fundamental task for the drug and material industry. Recent advances in graph neural networks (GNNs) have shown great promise in applying GNNs for molecular representation learning. Moreover, a few recent studies have also demonstrated successful applications of self-supervised learning methods to pre-train the GNNs to overcome the problem of insufficient labeled molecules. However, existing GNNs and pre-training strategies usually treat molecules as topological graph data without fully utilizing the molecular geometry information. Whereas, the three-dimensional (3D) spatial structure of a molecule, a.k.a molecular geometry, is one of the most critical factors for determining molecular physical, chemical, and biological properties. To this end, we propose a novel Geometry Enhanced Molecular representation learning method (GEM) for Chemical Representation Learning (ChemRL). At first, we design a geometry-based GNN architecture that simultaneously models atoms, bonds, and bond angles in a molecule. To be specific, we devised double graphs for a molecule: The first one encodes the atom-bond relations; The second one encodes bond-angle relations. Moreover, on top of the devised GNN architecture, we propose several novel geometry-level self-supervised learning strategies to learn spatial knowledge by utilizing the local and global molecular 3D structures. We compare ChemRL-GEM with various state-of-the-art (SOTA) baselines on different molecular benchmarks and exhibit that ChemRL-GEM can significantly outperform all baselines in both regression and classification tasks. For example, the experimental results show an overall improvement of 8.8% on average compared to SOTA baselines on the regression tasks, demonstrating the superiority of the proposed method.
LiteGEM: Lite Geometry Enhanced Molecular Representation Learning for Quantum Property Prediction
Jun 28, 2021

In this report, we (SuperHelix team) present our solution to KDD Cup 2021-PCQM4M-LSC, a large-scale quantum chemistry dataset on predicting HOMO-LUMO gap of molecules. Our solution, Lite Geometry Enhanced Molecular representation learning (LiteGEM) achieves a mean absolute error (MAE) of 0.1204 on the test set with the help of deep graph neural networks and various self-supervised learning tasks. The code of the framework can be found in https://github.com/PaddlePaddle/PaddleHelix/tree/dev/competition/kddcup2021-PCQM4M-LSC/.