 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeERNIE 5.0 Technical Report
Feb 04, 2026In this report, we introduce ERNIE 5.0, a natively autoregressive foundation model desinged for unified multimodal understanding and generation across text, image, video, and audio. All modalities are trained from scratch under a unified next-group-of-tokens prediction objective, based on an ultra-sparse mixture-of-experts (MoE) architecture with modality-agnostic expert routing. To address practical challenges in large-scale deployment under diverse resource constraints, ERNIE 5.0 adopts a novel elastic training paradigm. Within a single pre-training run, the model learns a family of sub-models with varying depths, expert capacities, and routing sparsity, enabling flexible trade-offs among performance, model size, and inference latency in memory- or time-constrained scenarios. Moreover, we systematically address the challenges of scaling reinforcement learning to unified foundation models, thereby guaranteeing efficient and stable post-training under ultra-sparse MoE architectures and diverse multimodal settings. Extensive experiments demonstrate that ERNIE 5.0 achieves strong and balanced performance across multiple modalities. To the best of our knowledge, among publicly disclosed models, ERNIE 5.0 represents the first production-scale realization of a trillion-parameter unified autoregressive model that supports both multimodal understanding and generation. To facilitate further research, we present detailed visualizations of modality-agnostic expert routing in the unified model, alongside comprehensive empirical analysis of elastic training, aiming to offer profound insights to the community.
CORD: Bridging the Audio-Text Reasoning Gap via Weighted On-policy Cross-modal Distillation
Jan 23, 2026Large Audio Language Models (LALMs) have garnered significant research interest. Despite being built upon text-based large language models (LLMs), LALMs frequently exhibit a degradation in knowledge and reasoning capabilities. We hypothesize that this limitation stems from the failure of current training paradigms to effectively bridge the acoustic-semantic gap within the feature representation space. To address this challenge, we propose CORD, a unified alignment framework that performs online cross-modal self-distillation. Specifically, it aligns audio-conditioned reasoning with its text-conditioned counterpart within a unified model. Leveraging the text modality as an internal teacher, CORD performs multi-granularity alignment throughout the audio rollout process. At the token level, it employs on-policy reverse KL divergence with importance-aware weighting to prioritize early and semantically critical tokens. At the sequence level, CORD introduces a judge-based global reward to optimize complete reasoning trajectories via Group Relative Policy Optimization (GRPO). Empirical results across multiple benchmarks demonstrate that CORD consistently enhances audio-conditioned reasoning and substantially bridges the audio-text performance gap with only 80k synthetic training samples, validating the efficacy and data efficiency of our on-policy, multi-level cross-modal alignment approach.
MoE Adapter for Large Audio Language Models: Sparsity, Disentanglement, and Gradient-Conflict-Free
Jan 08, 2026Extending the input modality of Large Language Models~(LLMs) to the audio domain is essential for achieving comprehensive multimodal perception. However, it is well-known that acoustic information is intrinsically \textit{heterogeneous}, entangling attributes such as speech, music, and environmental context. Existing research is limited to a dense, parameter-shared adapter to model these diverse patterns, which induces \textit{gradient conflict} during optimization, as parameter updates required for distinct attributes contradict each other. To address this limitation, we introduce the \textit{\textbf{MoE-Adapter}}, a sparse Mixture-of-Experts~(MoE) architecture designed to decouple acoustic information. Specifically, it employs a dynamic gating mechanism that routes audio tokens to specialized experts capturing complementary feature subspaces while retaining shared experts for global context, thereby mitigating gradient conflicts and enabling fine-grained feature learning. Comprehensive experiments show that the MoE-Adapter achieves superior performance on both audio semantic and paralinguistic tasks, consistently outperforming dense linear baselines with comparable computational costs. Furthermore, we will release the related code and models to facilitate future research.
Pre-Training on Large-Scale Generated Docking Conformations with HelixDock to Unlock the Potential of Protein-ligand Structure Prediction Models
Oct 21, 2023Molecular docking, a pivotal computational tool for drug discovery, predicts the binding interactions between small molecules (ligands) and target proteins (receptors). Conventional physics-based docking tools, though widely used, face limitations in precision due to restricted conformational sampling and imprecise scoring functions. Recent endeavors have employed deep learning techniques to enhance docking accuracy, but their generalization remains a concern due to limited training data. Leveraging the success of extensive and diverse data in other domains, we introduce HelixDock, a novel approach for site-specific molecular docking. Hundreds of millions of binding poses are generated by traditional docking tools, encompassing diverse protein targets and small molecules. Our deep learning-based docking model, a SE(3)-equivariant network, is pre-trained with this large-scale dataset and then fine-tuned with a small number of precise receptor-ligand complex structures. Comparative analyses against physics-based and deep learning-based baseline methods highlight HelixDock's superiority, especially on challenging test sets. Our study elucidates the scaling laws of the pre-trained molecular docking models, showcasing consistent improvements with increased model parameters and pre-train data quantities. Harnessing the power of extensive and diverse generated data holds promise for advancing AI-driven drug discovery.
GEM-2: Next Generation Molecular Property Prediction Network with Many-body and Full-range Interaction Modeling
Aug 15, 2022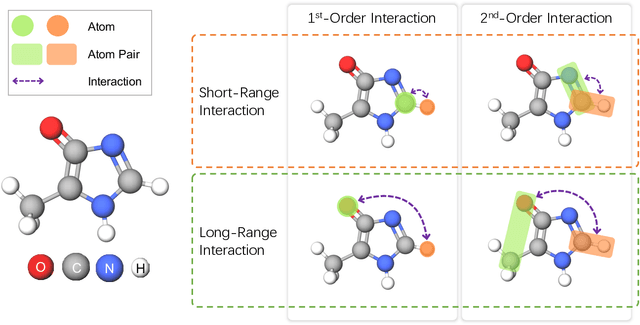
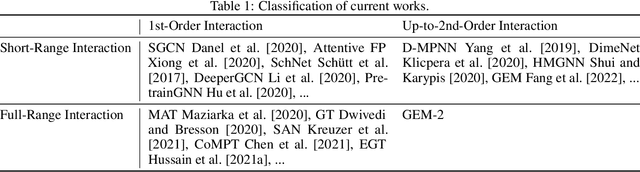
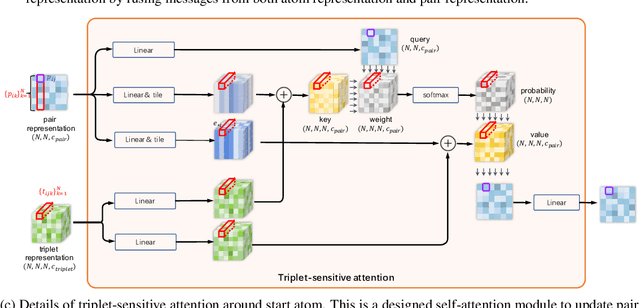
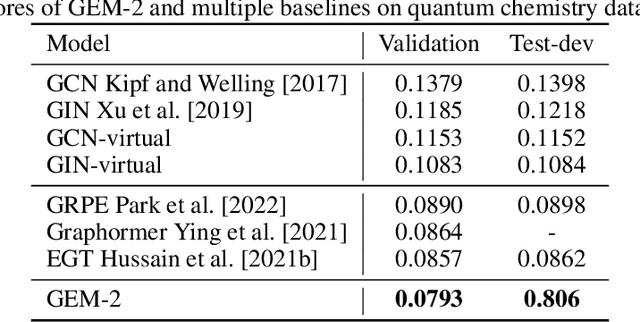
Molecular property prediction is a fundamental task in the drug and material industries. Physically, the properties of a molecule are determined by its own electronic structure, which can be exactly described by the Schr\"odinger equation. However, solving the Schr\"odinger equation for most molecules is extremely challenging due to long-range interactions in the behavior of a quantum many-body system. While deep learning methods have proven to be effective in molecular property prediction, we design a novel method, namely GEM-2, which comprehensively considers both the long-range and many-body interactions in molecules. GEM-2 consists of two interacted tracks: an atom-level track modeling both the local and global correlation between any two atoms, and a pair-level track modeling the correlation between all atom pairs, which embed information between any 3 or 4 atoms. Extensive experiments demonstrated the superiority of GEM-2 over multiple baseline methods in quantum chemistry and drug discovery tasks.
HelixFold-Single: MSA-free Protein Structure Prediction by Using Protein Language Model as an Alternative
Aug 09, 2022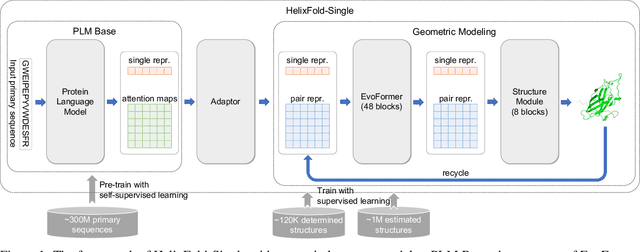


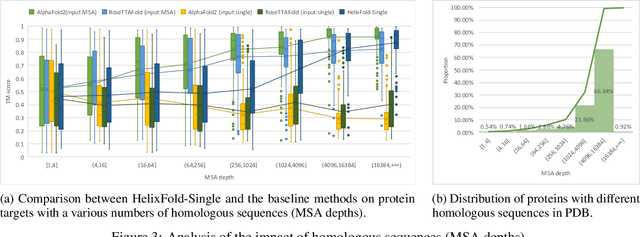
AI-based protein structure prediction pipelines, such as AlphaFold2, have achieved near-experimental accuracy. These advanced pipelines mainly rely on Multiple Sequence Alignments (MSAs) as inputs to learn the co-evolution information from the homologous sequences. Nonetheless, searching MSAs from protein databases is time-consuming, usually taking dozens of minutes. Consequently, we attempt to explore the limits of fast protein structure prediction by using only primary sequences of proteins. HelixFold-Single is proposed to combine a large-scale protein language model with the superior geometric learning capability of AlphaFold2. Our proposed method, HelixFold-Single, first pre-trains a large-scale protein language model (PLM) with thousands of millions of primary sequences utilizing the self-supervised learning paradigm, which will be used as an alternative to MSAs for learning the co-evolution information. Then, by combining the pre-trained PLM and the essential components of AlphaFold2, we obtain an end-to-end differentiable model to predict the 3D coordinates of atoms from only the primary sequence. HelixFold-Single is validated in datasets CASP14 and CAMEO, achieving competitive accuracy with the MSA-based methods on the targets with large homologous families. Furthermore, HelixFold-Single consumes much less time than the mainstream pipelines for protein structure prediction, demonstrating its potential in tasks requiring many predictions. The code of HelixFold-Single is available at https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold-single, and we also provide stable web services on https://paddlehelix.baidu.com/app/drug/protein-single/forecast.