 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEviAgent: Evidence-Driven Agent for Radiology Report Generation
Mar 14, 2026Automated radiology report generation holds immense potential to alleviate the heavy workload of radiologists. Despite the formidable vision-language capabilities of recent Multimodal Large Language Models (MLLMs), their clinical deployment is severely constrained by inherent limitations: their "black-box" decision-making renders the generated reports untraceable due to the lack of explicit visual evidence to support the diagnosis, and they struggle to access external domain knowledge. To address these challenges, we propose the Evidence-driven Radiology Report Generation Agent (EviAgent). Unlike opaque end-to-end paradigms, EviAgent coordinates a transparent reasoning trajectory by breaking down the complex generation process into granular operational units. We integrate multi-dimensional visual experts and retrieval mechanisms as external support modules, endowing the system with explicit visual evidence and high-quality clinical priors. Extensive experiments on MIMIC-CXR, CheXpert Plus, and IU-Xray datasets demonstrate that EviAgent outperforms both large-scale generalist models and specialized medical models, providing a robust and trustworthy solution for automated radiology report generation.
HelixFold-Single: MSA-free Protein Structure Prediction by Using Protein Language Model as an Alternative
Aug 09, 2022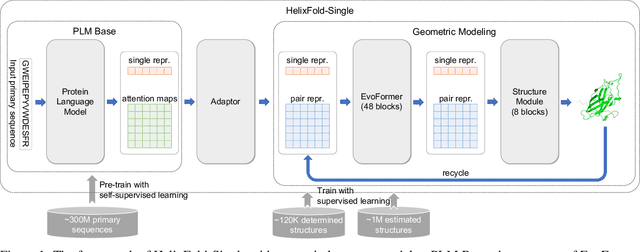


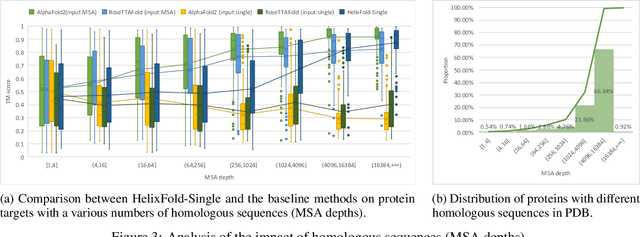
AI-based protein structure prediction pipelines, such as AlphaFold2, have achieved near-experimental accuracy. These advanced pipelines mainly rely on Multiple Sequence Alignments (MSAs) as inputs to learn the co-evolution information from the homologous sequences. Nonetheless, searching MSAs from protein databases is time-consuming, usually taking dozens of minutes. Consequently, we attempt to explore the limits of fast protein structure prediction by using only primary sequences of proteins. HelixFold-Single is proposed to combine a large-scale protein language model with the superior geometric learning capability of AlphaFold2. Our proposed method, HelixFold-Single, first pre-trains a large-scale protein language model (PLM) with thousands of millions of primary sequences utilizing the self-supervised learning paradigm, which will be used as an alternative to MSAs for learning the co-evolution information. Then, by combining the pre-trained PLM and the essential components of AlphaFold2, we obtain an end-to-end differentiable model to predict the 3D coordinates of atoms from only the primary sequence. HelixFold-Single is validated in datasets CASP14 and CAMEO, achieving competitive accuracy with the MSA-based methods on the targets with large homologous families. Furthermore, HelixFold-Single consumes much less time than the mainstream pipelines for protein structure prediction, demonstrating its potential in tasks requiring many predictions. The code of HelixFold-Single is available at https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold-single, and we also provide stable web services on https://paddlehelix.baidu.com/app/drug/protein-single/forecast.
HelixFold: An Efficient Implementation of AlphaFold2 using PaddlePaddle
Jul 13, 2022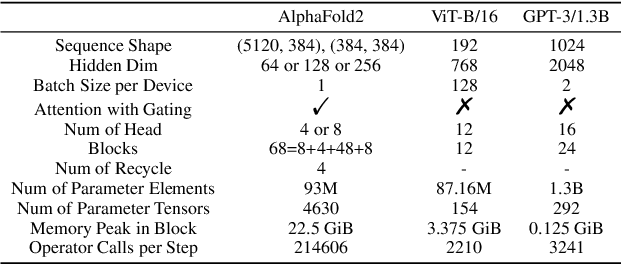
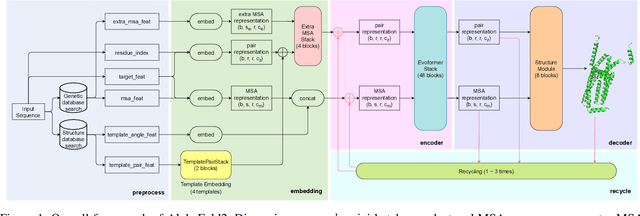
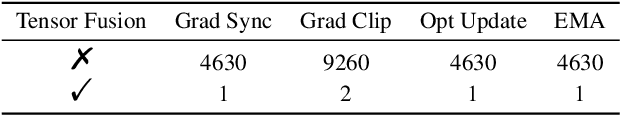
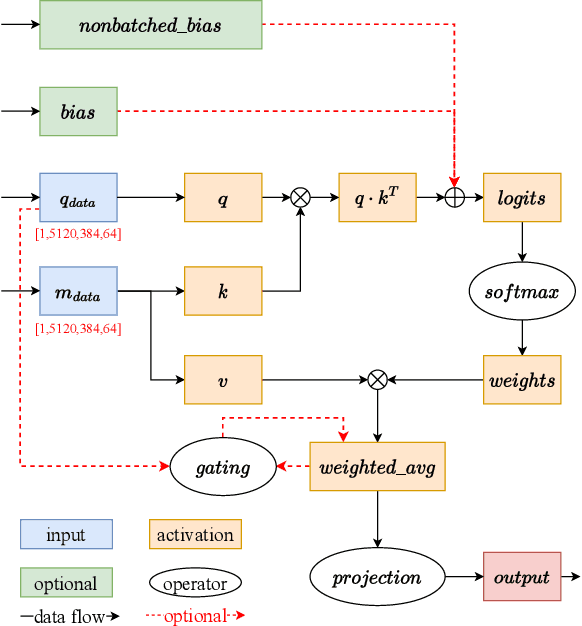
Accurate protein structure prediction can significantly accelerate the development of life science. The accuracy of AlphaFold2, a frontier end-to-end structure prediction system, is already close to that of the experimental determination techniques. Due to the complex model architecture and large memory consumption, it requires lots of computational resources and time to implement the training and inference of AlphaFold2 from scratch. The cost of running the original AlphaFold2 is expensive for most individuals and institutions. Therefore, reducing this cost could accelerate the development of life science. We implement AlphaFold2 using PaddlePaddle, namely HelixFold, to improve training and inference speed and reduce memory consumption. The performance is improved by operator fusion, tensor fusion, and hybrid parallelism computation, while the memory is optimized through Recompute, BFloat16, and memory read/write in-place. Compared with the original AlphaFold2 (implemented with Jax) and OpenFold (implemented with PyTorch), HelixFold needs only 7.5 days to complete the full end-to-end training and only 5.3 days when using hybrid parallelism, while both AlphaFold2 and OpenFold take about 11 days. HelixFold saves 1x training time. We verified that HelixFold's accuracy could be on par with AlphaFold2 on the CASP14 and CAMEO datasets. HelixFold's code is available on GitHub for free download: https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold, and we also provide stable web services on https://paddlehelix.baidu.com/app/drug/protein/forecast.