 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnd-to-End Reverse Screening Identifies Protein Targets of Small Molecules Using HelixFold3
Jan 20, 2026Identifying protein targets for small molecules, or reverse screening, is essential for understanding drug action, guiding compound repurposing, predicting off-target effects, and elucidating the molecular mechanisms of bioactive compounds. Despite its critical role, reverse screening remains challenging because accurately capturing interactions between a small molecule and structurally diverse proteins is inherently complex, and conventional step-wise workflows often propagate errors across decoupled steps such as target structure modeling, pocket identification, docking, and scoring. Here, we present an end-to-end reverse screening strategy leveraging HelixFold3, a high-accuracy biomolecular structure prediction model akin to AlphaFold3, which simultaneously models the folding of proteins from a protein library and the docking of small-molecule ligands within a unified framework. We validate this approach on a diverse and representative set of approximately one hundred small molecules. Compared with conventional reverse docking, our method improves screening accuracy and demonstrates enhanced structural fidelity, binding-site precision, and target prioritization. By systematically linking small molecules to their protein targets, this framework establishes a scalable and straightforward platform for dissecting molecular mechanisms, exploring off-target interactions, and supporting rational drug discovery.
Technical Report of HelixFold3 for Biomolecular Structure Prediction
Aug 30, 2024



The AlphaFold series has transformed protein structure prediction with remarkable accuracy, often matching experimental methods. AlphaFold2, AlphaFold-Multimer, and the latest AlphaFold3 represent significant strides in predicting single protein chains, protein complexes, and biomolecular structures. While AlphaFold2 and AlphaFold-Multimer are open-sourced, facilitating rapid and reliable predictions, AlphaFold3 remains partially accessible through a limited online server and has not been open-sourced, restricting further development. To address these challenges, the PaddleHelix team is developing HelixFold3, aiming to replicate AlphaFold3's capabilities. Using insights from previous models and extensive datasets, HelixFold3 achieves an accuracy comparable to AlphaFold3 in predicting the structures of conventional ligands, nucleic acids, and proteins. The initial release of HelixFold3 is available as open source on GitHub for academic research, promising to advance biomolecular research and accelerate discoveries. We also provide online service at PaddleHelix website at https://paddlehelix.baidu.com/app/all/helixfold3/forecast.
Pre-Training on Large-Scale Generated Docking Conformations with HelixDock to Unlock the Potential of Protein-ligand Structure Prediction Models
Oct 21, 2023Molecular docking, a pivotal computational tool for drug discovery, predicts the binding interactions between small molecules (ligands) and target proteins (receptors). Conventional physics-based docking tools, though widely used, face limitations in precision due to restricted conformational sampling and imprecise scoring functions. Recent endeavors have employed deep learning techniques to enhance docking accuracy, but their generalization remains a concern due to limited training data. Leveraging the success of extensive and diverse data in other domains, we introduce HelixDock, a novel approach for site-specific molecular docking. Hundreds of millions of binding poses are generated by traditional docking tools, encompassing diverse protein targets and small molecules. Our deep learning-based docking model, a SE(3)-equivariant network, is pre-trained with this large-scale dataset and then fine-tuned with a small number of precise receptor-ligand complex structures. Comparative analyses against physics-based and deep learning-based baseline methods highlight HelixDock's superiority, especially on challenging test sets. Our study elucidates the scaling laws of the pre-trained molecular docking models, showcasing consistent improvements with increased model parameters and pre-train data quantities. Harnessing the power of extensive and diverse generated data holds promise for advancing AI-driven drug discovery.
PASH at TREC 2021 Deep Learning Track: Generative Enhanced Model for Multi-stage Ranking
May 24, 2022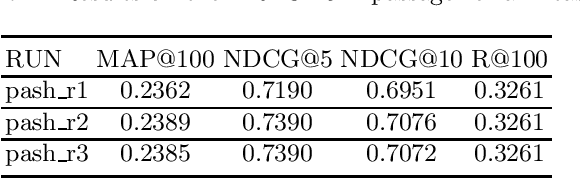
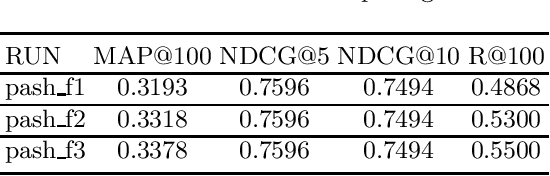
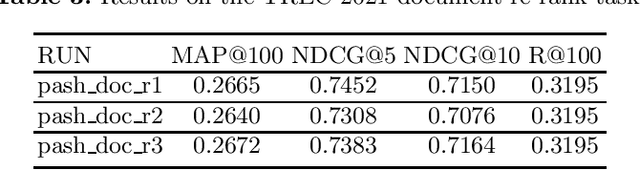
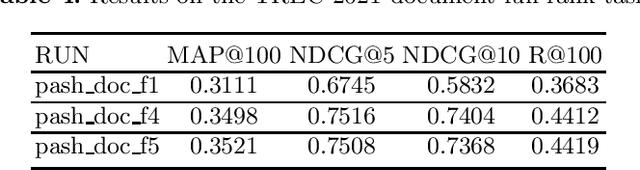
This paper describes the PASH participation in TREC 2021 Deep Learning Track. In the recall stage, we adopt a scheme combining sparse and dense retrieval method. In the multi-stage ranking phase, point-wise and pair-wise ranking strategies are used one after another based on model continual pre-trained on general knowledge and document-level data. Compared to TREC 2020 Deep Learning Track, we have additionally introduced the generative model T5 to further enhance the performance.
CandidateDrug4Cancer: An Open Molecular Graph Learning Benchmark on Drug Discovery for Cancer
Mar 02, 2022

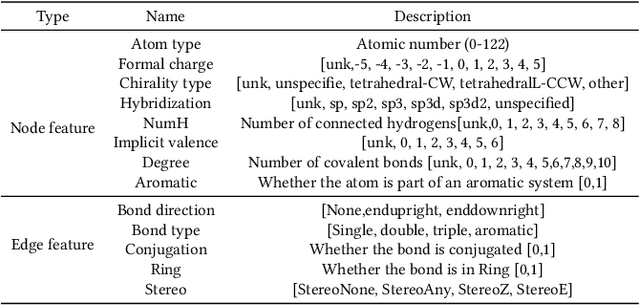
Anti-cancer drug discoveries have been serendipitous, we sought to present the Open Molecular Graph Learning Benchmark, named CandidateDrug4Cancer, a challenging and realistic benchmark dataset to facilitate scalable, robust, and reproducible graph machine learning research for anti-cancer drug discovery. CandidateDrug4Cancer dataset encompasses multiple most-mentioned 29 targets for cancer, covering 54869 cancer-related drug molecules which are ranged from pre-clinical, clinical and FDA-approved. Besides building the datasets, we also perform benchmark experiments with effective Drug Target Interaction (DTI) prediction baselines using descriptors and expressive graph neural networks. Experimental results suggest that CandidateDrug4Cancer presents significant challenges for learning molecular graphs and targets in practical application, indicating opportunities for future researches on developing candidate drugs for treating cancers.
Docking-based Virtual Screening with Multi-Task Learning
Nov 18, 2021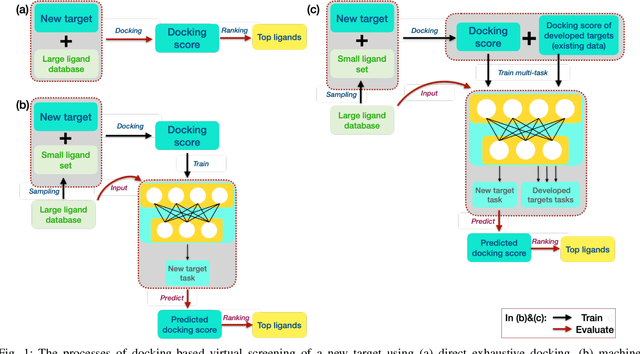
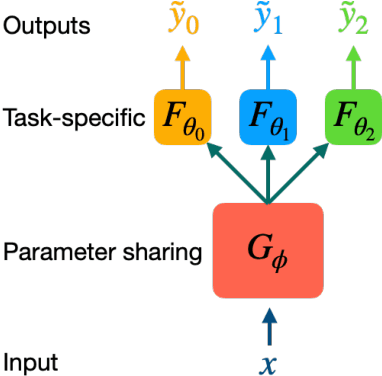
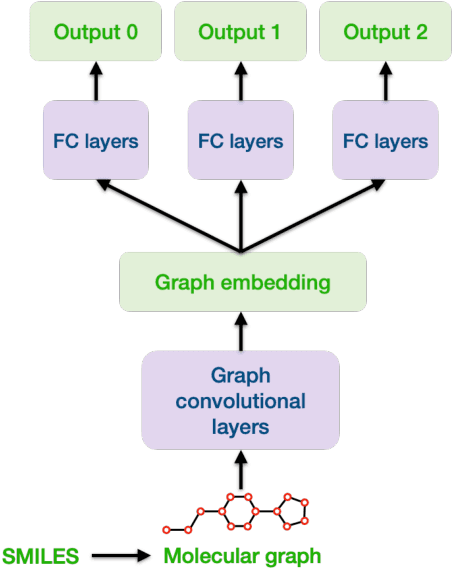
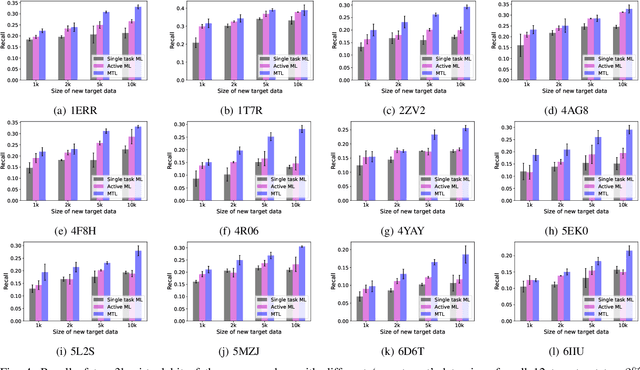
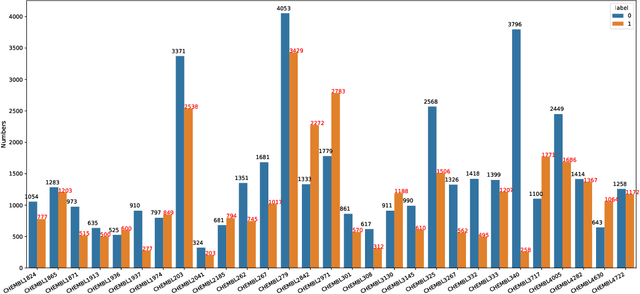
Machine learning shows great potential in virtual screening for drug discovery. Current efforts on accelerating docking-based virtual screening do not consider using existing data of other previously developed targets. To make use of the knowledge of the other targets and take advantage of the existing data, in this work, we apply multi-task learning to the problem of docking-based virtual screening. With two large docking datasets, the results of extensive experiments show that multi-task learning can achieve better performances on docking score prediction. By learning knowledge across multiple targets, the model trained by multi-task learning shows a better ability to adapt to a new target. Additional empirical study shows that other problems in drug discovery, such as the experimental drug-target affinity prediction, may also benefit from multi-task learning. Our results demonstrate that multi-task learning is a promising machine learning approach for docking-based virtual screening and accelerating the process of drug discovery.
Winner Team Mia at TextVQA Challenge 2021: Vision-and-Language Representation Learning with Pre-trained Sequence-to-Sequence Model
Jun 24, 2021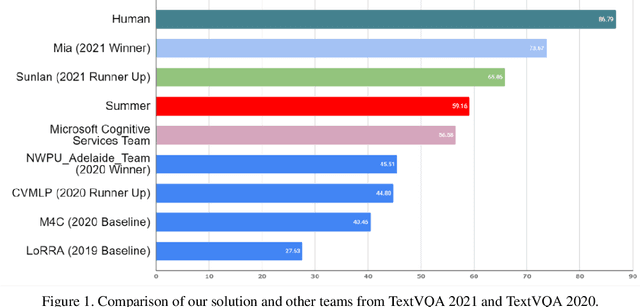
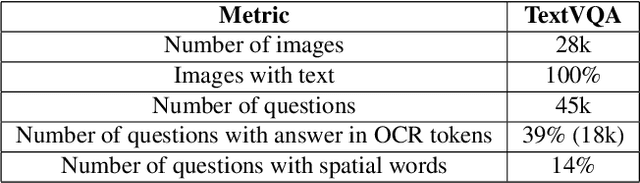
TextVQA requires models to read and reason about text in images to answer questions about them. Specifically, models need to incorporate a new modality of text present in the images and reason over it to answer TextVQA questions. In this challenge, we use generative model T5 for TextVQA task. Based on pre-trained checkpoint T5-3B from HuggingFace repository, two other pre-training tasks including masked language modeling(MLM) and relative position prediction(RPP) are designed to better align object feature and scene text. In the stage of pre-training, encoder is dedicate to handle the fusion among multiple modalities: question text, object text labels, scene text labels, object visual features, scene visual features. After that decoder generates the text sequence step-by-step, cross entropy loss is required by default. We use a large-scale scene text dataset in pre-training and then fine-tune the T5-3B with the TextVQA dataset only.