 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgentTrap: Measuring Runtime Trust Failures in Third-Party Agent Skills
May 13, 2026Third-party skills are becoming the package ecosystem for LLM agents. They package natural-language instructions, helper scripts, templates, documents, and service configuration into reusable workflows. This makes skills useful, but it also introduces a new security problem: a malicious skill does not need to ask the model to perform an obviously harmful action. Instead, it can disguise the harmful behavior as part of a routine workflow, relying on the agent to execute that workflow with high-value permissions and limited human supervision. We introduce AgentTrap, a dynamic benchmark for evaluating whether LLM agents can use third-party skills while resisting malicious runtime behavior. AgentTrap contains 141 tasks: 91 malicious tasks and 50 benign utility tasks, covering 16 security-impact dimensions grounded in agent-skill supply-chain threats. In each task, the agent receives an ordinary user request, runs with installed skills that may contain malicious workflow elements, and is executed in a sandboxed environment. AgentTrap then judges complete trajectories for attack success, blocked or refused behavior, attack-not-triggered cases, and no-attack-evidence outcomes. Our central finding is that the most informative failures are not simple jailbreaks. Models often complete the visible user task while treating unsafe side effects introduced by the skill as part of the normal workflow. This motivates runtime evaluation of the concrete model--framework--workspace environment in which users actually delegate work. Code and data are available at https://github.com/zhmzm/AgentTrap and https://huggingface.co/datasets/zhmzm/AgentTrap.
Transferable FB-GNN-MBE Framework for Potential Energy Surfaces: Data-Adaptive Transfer Learning in Deep Learned Many-Body Expansion Theory
Apr 10, 2026Mechanistic understanding and rational design of complex chemical systems depend on fast and accurate predictions of electronic structures beyond individual building blocks. However, if the system exceeds hundreds of atoms, first-principles quantum mechanical (QM) modeling becomes impractical. In this study, we developed FB-GNN-MBE by integrating a fragment-based graph neural network (FB-GNN) into the many-body expansion (MBE) theory and demonstrated its capacity to reproduce first-principles potential energy surfaces (PES) for hierarchically structured systems with manageable accuracy, complexity, and interpretability. Specifically, we divided the entire system into basic building blocks (fragments), evaluated their one-fragment energies using a QM model, and addressed many-fragment interactions using the structure-property relationships trained by FB-GNNs. Our investigation shows that FB-GNN-MBE achieves chemical accuracy in predicting two-body (2B) and three-body (3B) energies across water, phenol, and mixture benchmarks, as well as the one-dimensional dissociation curves of water and phenol dimers. To transfer the success of FB-GNN-MBE across various systems with minimal computational costs and data demands, we developed and validated a teacher-student learning protocol. A heavy-weight FB-GNN trained on a mixed-density water cluster ensemble (teacher) distills its learned knowledge and passes it to a light-weight GNN (student), which is later fine-tuned on a uniform-density (H2O)21 cluster ensemble. This transfer learning strategy resulted in efficient and accurate prediction of 2B and 3B energies for variously sized water clusters without retraining. Our transferable FB-GNN-MBE framework outperformed conventional non-FB-GNN-based models and showed high practicality for large-scale molecular simulations.
SenseMath: Do LLMs Have Number Sense? Evaluating Shortcut Use, Judgment, and Generation
Apr 02, 2026Large language models often default to step-by-step computation even when efficient numerical shortcuts are available. This raises a basic question: do they exhibit number sense in a human-like behavioral sense, i.e., the ability to recognize numerical structure, apply shortcuts when appropriate, and avoid them when they are not? We introduce SenseMath, a controlled benchmark for evaluating structure-sensitive numerical reasoning in LLMs. SenseMath contains 4,800 items spanning eight shortcut categories and four digit scales, with matched strong-shortcut, weak-shortcut, and control variants. It supports three evaluation settings of increasing cognitive demand: Shortcut Use (whether models can apply shortcuts on shortcut-amenable problems); Applicability Judgment (whether they can recognize when a shortcut is appropriate or misleading); and Problem Generation (whether they can generate new problem items that correctly admit a given type of shortcut). Our evaluation across five LLMs, ranging from GPT-4o-mini to Llama-3.1-8B, shows a consistent pattern: when explicitly prompted, models readily adopt shortcut strategies and achieve substantial accuracy gains on shortcut-amenable items (up to 15%), yet under standard chain-of-thought prompting they spontaneously employ such strategies in fewer than 40% of cases, even when they demonstrably possess the requisite capability. Moreover, this competence is confined to the Use level; models systematically over-generalise shortcuts to problems where they do not apply, and fail to generate valid shortcut-bearing problems from scratch. Together, these results suggest that current LLMs exhibit procedural shortcut fluency without the structural understanding of when and why shortcuts work that underlies human number sense.
Driving Reaction Trajectories via Latent Flow Matching
Feb 11, 2026Recent advances in reaction prediction have achieved near-saturated accuracy on standard benchmarks (e.g., USPTO), yet most state-of-the-art models formulate the task as a one-shot mapping from reactants to products, offering limited insight into the underlying reaction process. Procedural alternatives introduce stepwise generation but often rely on mechanism-specific supervision, discrete symbolic edits, and computationally expensive inference. In this work, we propose LatentRxnFlow, a new reaction prediction paradigm that models reactions as continuous latent trajectories anchored at the thermodynamic product state. Built on Conditional Flow Matching, our approach learns time-dependent latent dynamics directly from standard reactant-product pairs, without requiring mechanistic annotations or curated intermediate labels. While LatentRxnFlow achieves state-of-the-art performance on USPTO benchmarks, more importantly, the continuous formulation exposes the full generative trajectory, enabling trajectory-level diagnostics that are difficult to realize with discrete or one-shot models. We show that latent trajectory analysis allows us to localize and characterize failure modes and to mitigate certain errors via gated inference. Furthermore, geometric properties of the learned trajectories provide an intrinsic signal of epistemic uncertainty, helping prioritize reliably predictable reaction outcomes and flag ambiguous cases for additional validation. Overall, LatentRxnFlow combines strong predictive accuracy with improved transparency, diagnosability, and uncertainty awareness, moving reaction prediction toward more trustworthy deployment in high-throughput discovery workflows.
Integrating Graph Neural Networks and Many-Body Expansion Theory for Potential Energy Surfaces
Nov 03, 2024



Rational design of next-generation functional materials relied on quantitative predictions of their electronic structures beyond single building blocks. First-principles quantum mechanical (QM) modeling became infeasible as the size of a material grew beyond hundreds of atoms. In this study, we developed a new computational tool integrating fragment-based graph neural networks (FBGNN) into the fragment-based many-body expansion (MBE) theory, referred to as FBGNN-MBE, and demonstrated its capacity to reproduce full-dimensional potential energy surfaces (FD-PES) for hierarchic chemical systems with manageable accuracy, complexity, and interpretability. In particular, we divided the entire system into basic building blocks (fragments), evaluated their single-fragment energies using a first-principles QM model and attacked many-fragment interactions using the structure-property relationships trained by FBGNNs. Our development of FBGNN-MBE demonstrated the potential of a new framework integrating deep learning models into fragment-based QM methods, and marked a significant step towards computationally aided design of large functional materials.
TCR: A Transformer Based Deep Network for Predicting Cancer Drugs Response
Jul 10, 2022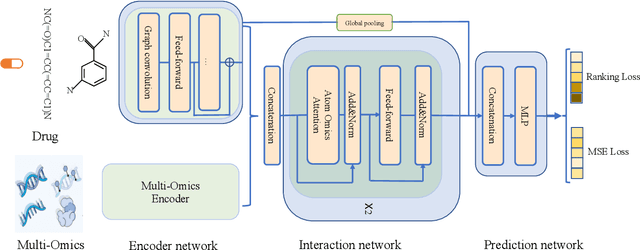
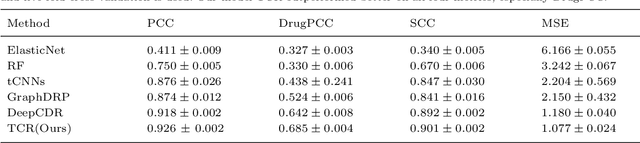
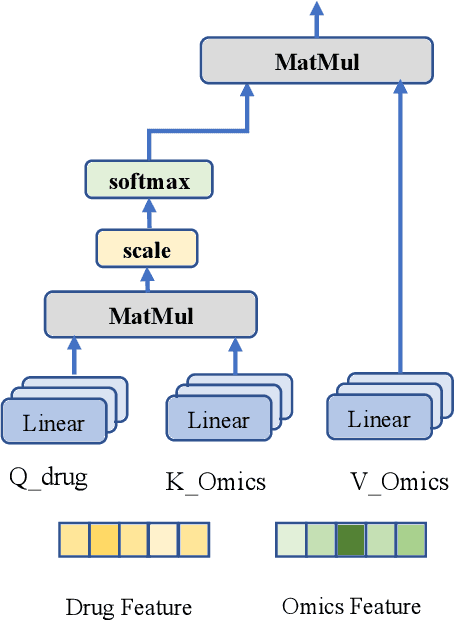

Predicting clinical outcomes to anti-cancer drugs on a personalized basis is challenging in cancer treatment due to the heterogeneity of tumors. Traditional computational efforts have been made to model the effect of drug response on individual samples depicted by their molecular profile, yet overfitting occurs because of the high dimension for omics data, hindering models from clinical application. Recent research shows that deep learning is a promising approach to build drug response models by learning alignment patterns between drugs and samples. However, existing studies employed the simple feature fusion strategy and only considered the drug features as a whole representation while ignoring the substructure information that may play a vital role when aligning drugs and genes. Hereby in this paper, we propose TCR (Transformer based network for Cancer drug Response) to predict anti-cancer drug response. By utilizing an attention mechanism, TCR is able to learn the interactions between drug atom/sub-structure and molecular signatures efficiently in our study. Furthermore, a dual loss function and cross sampling strategy were designed to improve the prediction power of TCR. We show that TCR outperformed all other methods under various data splitting strategies on all evaluation matrices (some with significant improvement). Extensive experiments demonstrate that TCR shows significantly improved generalization ability on independent in-vitro experiments and in-vivo real patient data. Our study highlights the prediction power of TCR and its potential value for cancer drug repurpose and precision oncology treatment.
Improving Subgraph Representation Learning via Multi-View Augmentation
May 25, 2022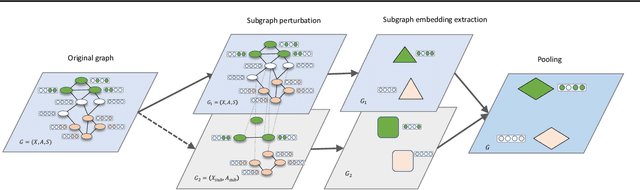
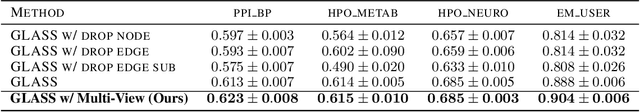
Subgraph representation learning based on Graph Neural Network (GNN) has broad applications in chemistry and biology, such as molecule property prediction and gene collaborative function prediction. On the other hand, graph augmentation techniques have shown promising results in improving graph-based and node-based classification tasks but are rarely explored in the GNN-based subgraph representation learning literature. In this work, we developed a novel multiview augmentation mechanism to improve subgraph representation learning and thus the accuracy of downstream prediction tasks. The augmentation technique creates multiple variants of subgraphs and embeds these variants into the original graph to achieve both high training efficiency, scalability, and improved accuracy. Experiments on several real-world subgraph benchmarks demonstrate the superiority of our proposed multi-view augmentation techniques.