 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHumanizing Robot Gaze Shifts: A Framework for Natural Gaze Shifts in Humanoid Robots
Feb 25, 2026Leveraging auditory and visual feedback for attention reorientation is essential for natural gaze shifts in social interaction. However, enabling humanoid robots to perform natural and context-appropriate gaze shifts in unconstrained human--robot interaction (HRI) remains challenging, as it requires the coupling of cognitive attention mechanisms and biomimetic motion generation. In this work, we propose the Robot Gaze-Shift (RGS) framework, which integrates these two components into a unified pipeline. First, RGS employs a vision--language model (VLM)-based gaze reasoning pipeline to infer context-appropriate gaze targets from multimodal interaction cues, ensuring consistency with human gaze-orienting regularities. Second, RGS introduces a conditional Vector Quantized-Variational Autoencoder (VQ-VAE) model for eye--head coordinated gaze-shift motion generation, producing diverse and human-like gaze-shift behaviors. Experiments validate that RGS effectively replicates human-like target selection and generates realistic, diverse gaze-shift motions.
Leveraging Diffusion Knowledge for Generative Image Compression with Fractal Frequency-Aware Band Learning
Mar 14, 2025By optimizing the rate-distortion-realism trade-off, generative image compression approaches produce detailed, realistic images instead of the only sharp-looking reconstructions produced by rate-distortion-optimized models. In this paper, we propose a novel deep learning-based generative image compression method injected with diffusion knowledge, obtaining the capacity to recover more realistic textures in practical scenarios. Efforts are made from three perspectives to navigate the rate-distortion-realism trade-off in the generative image compression task. First, recognizing the strong connection between image texture and frequency-domain characteristics, we design a Fractal Frequency-Aware Band Image Compression (FFAB-IC) network to effectively capture the directional frequency components inherent in natural images. This network integrates commonly used fractal band feature operations within a neural non-linear mapping design, enhancing its ability to retain essential given information and filter out unnecessary details. Then, to improve the visual quality of image reconstruction under limited bandwidth, we integrate diffusion knowledge into the encoder and implement diffusion iterations into the decoder process, thus effectively recovering lost texture details. Finally, to fully leverage the spatial and frequency intensity information, we incorporate frequency- and content-aware regularization terms to regularize the training of the generative image compression network. Extensive experiments in quantitative and qualitative evaluations demonstrate the superiority of the proposed method, advancing the boundaries of achievable distortion-realism pairs, i.e., our method achieves better distortions at high realism and better realism at low distortion than ever before.
SHREC 2021: Classification in cryo-electron tomograms
Mar 18, 2022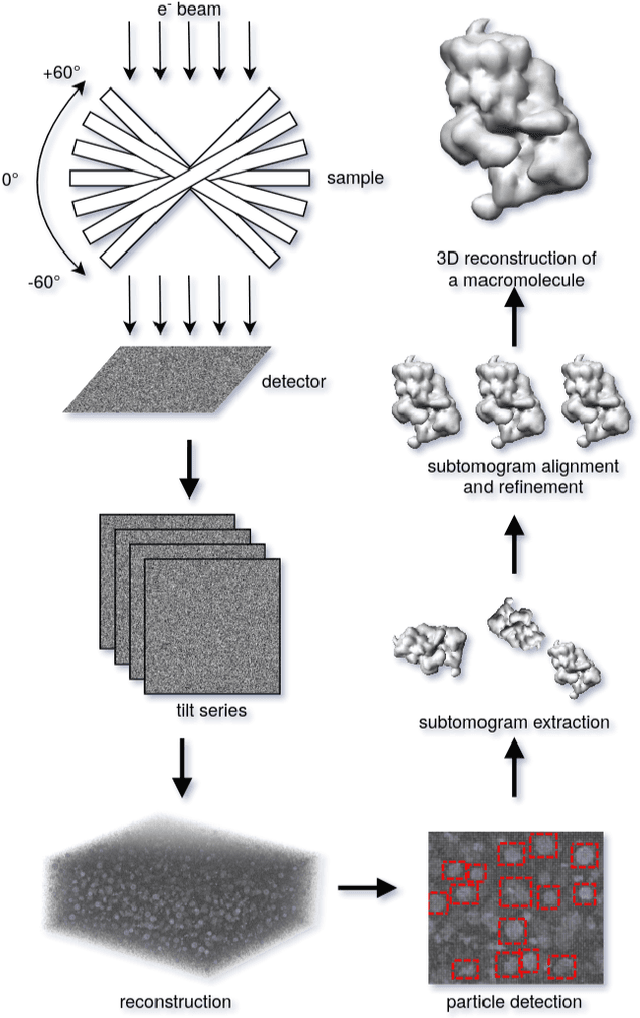
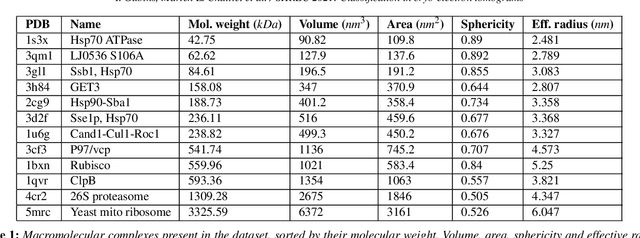
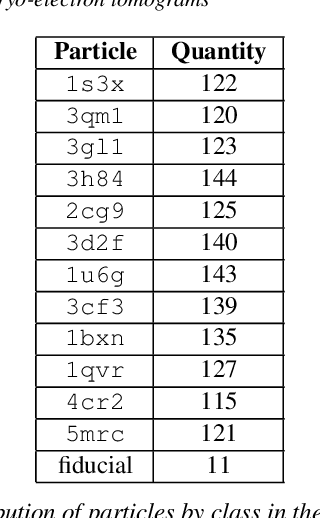
Cryo-electron tomography (cryo-ET) is an imaging technique that allows three-dimensional visualization of macro-molecular assemblies under near-native conditions. Cryo-ET comes with a number of challenges, mainly low signal-to-noise and inability to obtain images from all angles. Computational methods are key to analyze cryo-electron tomograms. To promote innovation in computational methods, we generate a novel simulated dataset to benchmark different methods of localization and classification of biological macromolecules in tomograms. Our publicly available dataset contains ten tomographic reconstructions of simulated cell-like volumes. Each volume contains twelve different types of complexes, varying in size, function and structure. In this paper, we have evaluated seven different methods of finding and classifying proteins. Seven research groups present results obtained with learning-based methods and trained on the simulated dataset, as well as a baseline template matching (TM), a traditional method widely used in cryo-ET research. We show that learning-based approaches can achieve notably better localization and classification performance than TM. We also experimentally confirm that there is a negative relationship between particle size and performance for all methods.
Boosting Active Learning via Improving Test Performance
Dec 10, 2021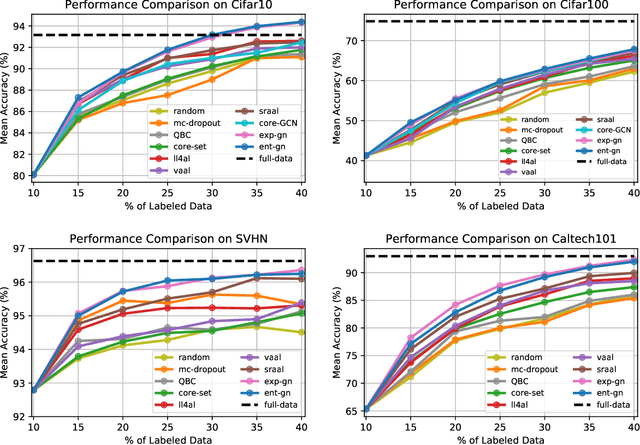
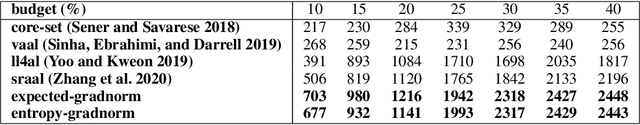
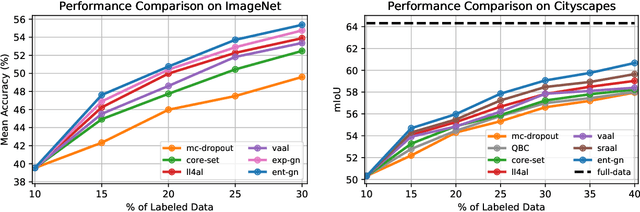
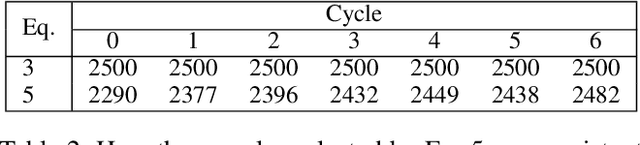
Central to active learning (AL) is what data should be selected for annotation. Existing works attempt to select highly uncertain or informative data for annotation. Nevertheless, it remains unclear how selected data impacts the test performance of the task model used in AL. In this work, we explore such an impact by theoretically proving that selecting unlabeled data of higher gradient norm leads to a lower upper bound of test loss, resulting in a better test performance. However, due to the lack of label information, directly computing gradient norm for unlabeled data is infeasible. To address this challenge, we propose two schemes, namely expected-gradnorm and entropy-gradnorm. The former computes the gradient norm by constructing an expected empirical loss while the latter constructs an unsupervised loss with entropy. Furthermore, we integrate the two schemes in a universal AL framework. We evaluate our method on classical image classification and semantic segmentation tasks. To demonstrate its competency in domain applications and its robustness to noise, we also validate our method on a cellular imaging analysis task, namely cryo-Electron Tomography subtomogram classification. Results demonstrate that our method achieves superior performance against the state-of-the-art. Our source code is available at https://github.com/xulabs/aitom
* 13 pages
Cryo-shift: Reducing domain shift in cryo-electron subtomograms with unsupervised domain adaptation and randomization
Nov 17, 2021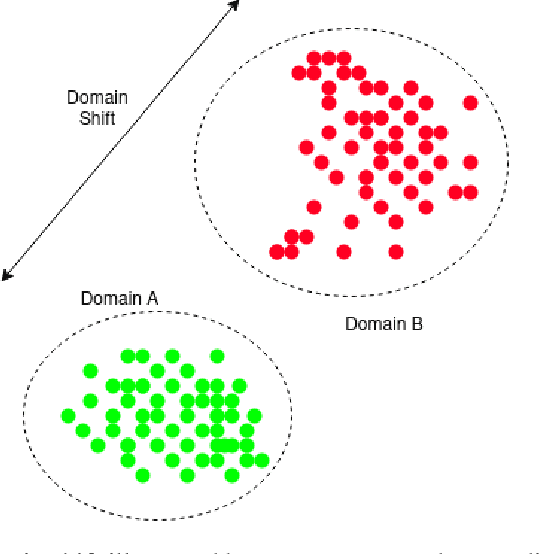

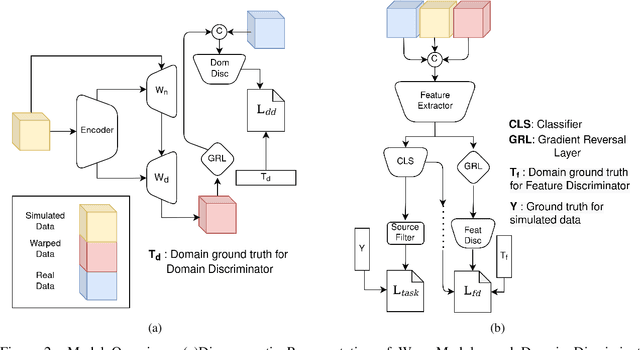

Cryo-Electron Tomography (cryo-ET) is a 3D imaging technology that enables the visualization of subcellular structures in situ at near-atomic resolution. Cellular cryo-ET images help in resolving the structures of macromolecules and determining their spatial relationship in a single cell, which has broad significance in cell and structural biology. Subtomogram classification and recognition constitute a primary step in the systematic recovery of these macromolecular structures. Supervised deep learning methods have been proven to be highly accurate and efficient for subtomogram classification, but suffer from limited applicability due to scarcity of annotated data. While generating simulated data for training supervised models is a potential solution, a sizeable difference in the image intensity distribution in generated data as compared to real experimental data will cause the trained models to perform poorly in predicting classes on real subtomograms. In this work, we present Cryo-Shift, a fully unsupervised domain adaptation and randomization framework for deep learning-based cross-domain subtomogram classification. We use unsupervised multi-adversarial domain adaption to reduce the domain shift between features of simulated and experimental data. We develop a network-driven domain randomization procedure with `warp' modules to alter the simulated data and help the classifier generalize better on experimental data. We do not use any labeled experimental data to train our model, whereas some of the existing alternative approaches require labeled experimental samples for cross-domain classification. Nevertheless, Cryo-Shift outperforms the existing alternative approaches in cross-domain subtomogram classification in extensive evaluation studies demonstrated herein using both simulated and experimental data.
* 14 pages
Disentangling semantic features of macromolecules in Cryo-Electron Tomography
Jun 27, 2021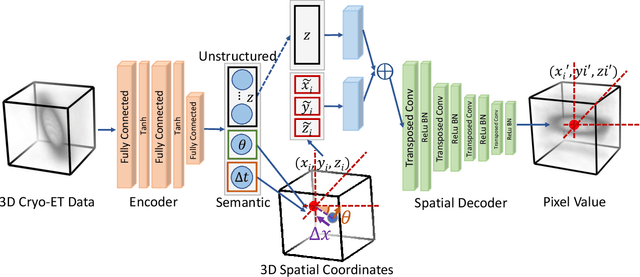
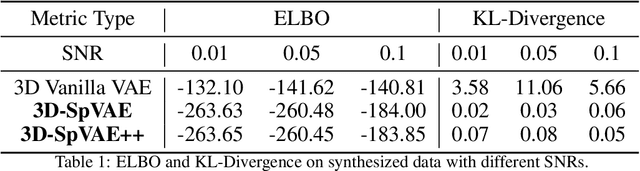
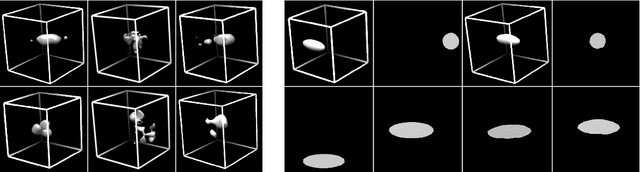

Cryo-electron tomography (Cryo-ET) is a 3D imaging technique that enables the systemic study of shape, abundance, and distribution of macromolecular structures in single cells in near-atomic resolution. However, the systematic and efficient $\textit{de novo}$ recognition and recovery of macromolecular structures captured by Cryo-ET are very challenging due to the structural complexity and imaging limits. Even macromolecules with identical structures have various appearances due to different orientations and imaging limits, such as noise and the missing wedge effect. Explicitly disentangling the semantic features of macromolecules is crucial for performing several downstream analyses on the macromolecules. This paper has addressed the problem by proposing a 3D Spatial Variational Autoencoder that explicitly disentangle the structure, orientation, and shift of macromolecules. Extensive experiments on both synthesized and real cryo-ET datasets and cross-domain evaluations demonstrate the efficacy of our method.
Active Learning to Classify Macromolecular Structures in situ for Less Supervision in Cryo-Electron Tomography
Feb 24, 2021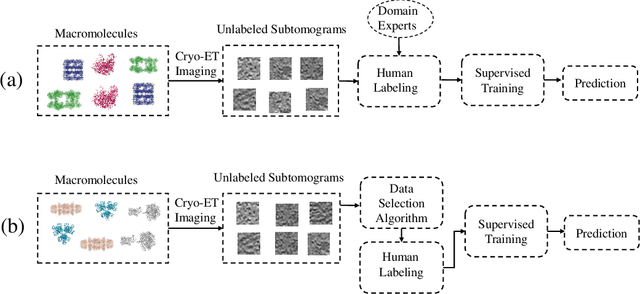
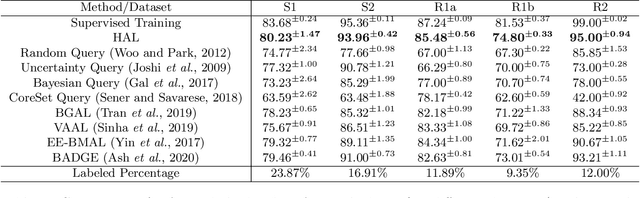
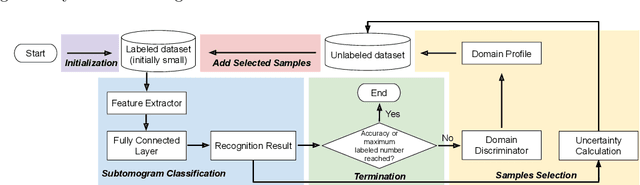
Motivation: Cryo-Electron Tomography (cryo-ET) is a 3D bioimaging tool that visualizes the structural and spatial organization of macromolecules at a near-native state in single cells, which has broad applications in life science. However, the systematic structural recognition and recovery of macromolecules captured by cryo-ET are difficult due to high structural complexity and imaging limits. Deep learning based subtomogram classification have played critical roles for such tasks. As supervised approaches, however, their performance relies on sufficient and laborious annotation on a large training dataset. Results: To alleviate this major labeling burden, we proposed a Hybrid Active Learning (HAL) framework for querying subtomograms for labelling from a large unlabeled subtomogram pool. Firstly, HAL adopts uncertainty sampling to select the subtomograms that have the most uncertain predictions. Moreover, to mitigate the sampling bias caused by such strategy, a discriminator is introduced to judge if a certain subtomogram is labeled or unlabeled and subsequently the model queries the subtomogram that have higher probabilities to be unlabeled. Additionally, HAL introduces a subset sampling strategy to improve the diversity of the query set, so that the information overlap is decreased between the queried batches and the algorithmic efficiency is improved. Our experiments on subtomogram classification tasks using both simulated and real data demonstrate that we can achieve comparable testing performance (on average only 3% accuracy drop) by using less than 30% of the labeled subtomograms, which shows a very promising result for subtomogram classification task with limited labeling resources.
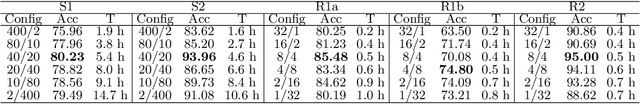
Feedback-Based Dynamic Feature Selection for Constrained Continuous Data Acquisition
Nov 10, 2020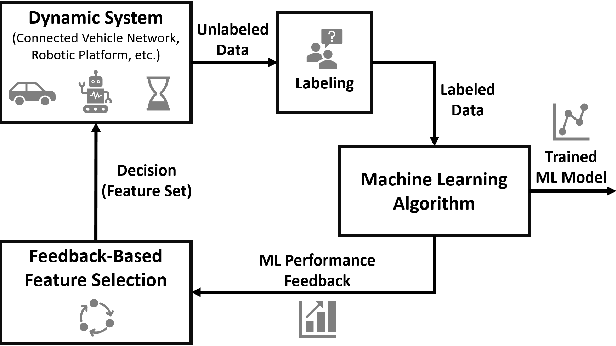
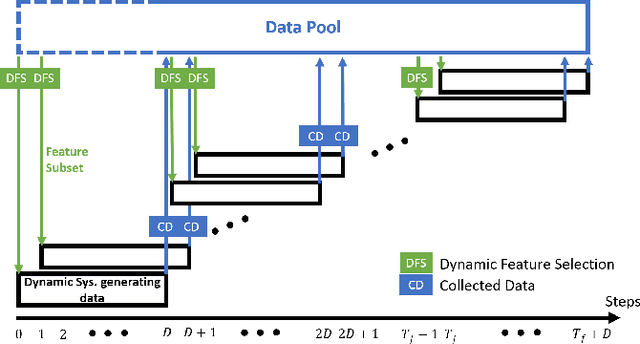
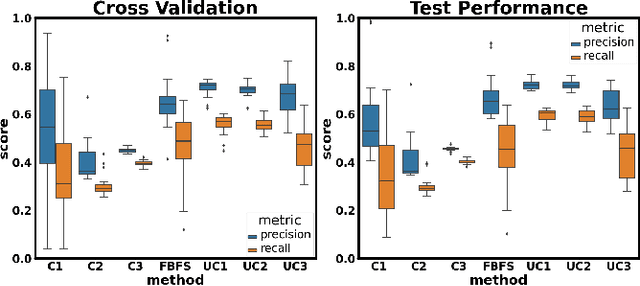

Relevant and high-quality data are critical to successful development of machine learning applications. For machine learning applications on dynamic systems equipped with a large number of sensors, such as connected vehicles and robots, how to find relevant and high-quality data features in an efficient way is a challenging problem. In this work, we address the problem of feature selection in constrained continuous data acquisition. We propose a feedback-based dynamic feature selection algorithm that efficiently decides on the feature set for data collection from a dynamic system in a step-wise manner. We formulate the sequential feature selection procedure as a Markov Decision Process. The machine learning model performance feedback with an exploration component is used as the reward function in an $\epsilon$-greedy action selection. Our evaluation shows that the proposed feedback-based feature selection algorithm has superior performance over constrained baseline methods and matching performance with unconstrained baseline methods.
Few shot domain adaptation for in situ macromolecule structural classification in cryo-electron tomograms
Jul 30, 2020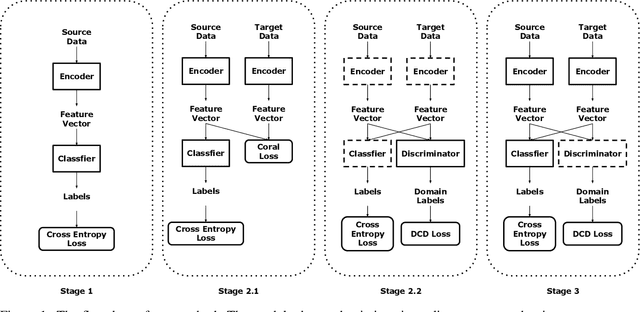
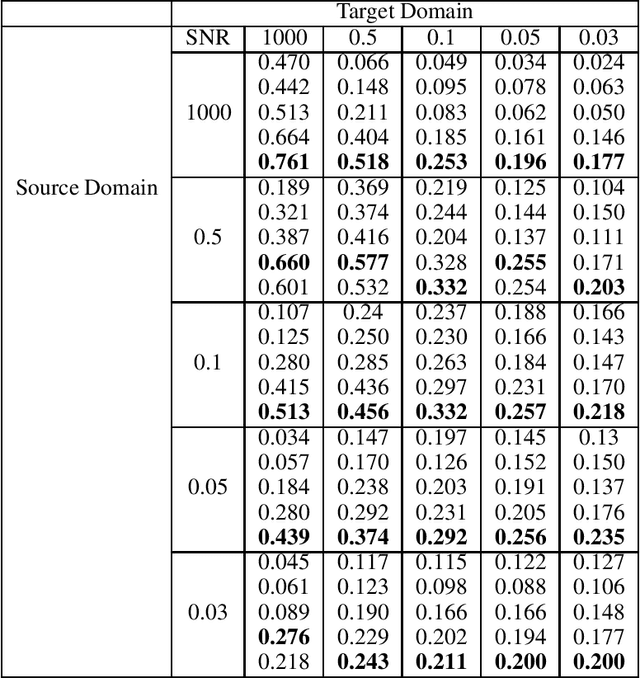
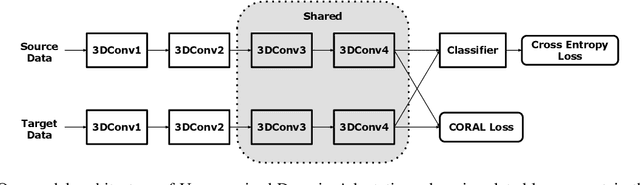

Motivation: Cryo-Electron Tomography (cryo-ET) visualizes structure and spatial organization of macromolecules and their interactions with other subcellular components inside single cells in the close-to-native state at sub-molecular resolution. Such information is critical for the accurate understanding of cellular processes. However, subtomogram classification remains one of the major challenges for the systematic recognition and recovery of the macromolecule structures in cryo-ET because of imaging limits and data quantity. Recently, deep learning has significantly improved the throughput and accuracy of large-scale subtomogram classification. However often it is difficult to get enough high-quality annotated subtomogram data for supervised training due to the enormous expense of labeling. To tackle this problem, it is beneficial to utilize another already annotated dataset to assist the training process. However, due to the discrepancy of image intensity distribution between source domain and target domain, the model trained on subtomograms in source domainmay perform poorly in predicting subtomogram classes in the target domain. Results: In this paper, we adapt a few shot domain adaptation method for deep learning based cross-domain subtomogram classification. The essential idea of our method consists of two parts: 1) take full advantage of the distribution of plentiful unlabeled target domain data, and 2) exploit the correlation between the whole source domain dataset and few labeled target domain data. Experiments conducted on simulated and real datasets show that our method achieves significant improvement on cross domain subtomogram classification compared with baseline methods.
* This article has been accepted for publication in Bioinformatics Published by Oxford University Press
AITom: Open-source AI platform for cryo-electron Tomography data analysis
Nov 08, 2019
Cryo-electron tomography (cryo-ET) is an emerging technology for the 3D visualization of structural organizations and interactions of subcellular components at near-native state and sub-molecular resolution. Tomograms captured by cryo-ET contain heterogeneous structures representing the complex and dynamic subcellular environment. Since the structures are not purified or fluorescently labeled, the spatial organization and interaction between both the known and unknown structures can be studied in their native environment. The rapid advances of cryo-electron tomography (cryo-ET) have generated abundant 3D cellular imaging data. However, the systematic localization, identification, segmentation, and structural recovery of the subcellular components require efficient and accurate large-scale image analysis methods. We introduce AITom, an open-source artificial intelligence platform for cryo-ET researchers. AITom provides many public as well as in-house algorithms for performing cryo-ET data analysis through both the traditional template-based or template-free approach and the deep learning approach. Comprehensive tutorials for each analysis module are provided to guide the user through. We welcome researchers and developers to join this collaborative open-source software development project. Availability: https://github.com/xulabs/aitom