 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSHREC 2021: Classification in cryo-electron tomograms
Paper and Code
Mar 18, 2022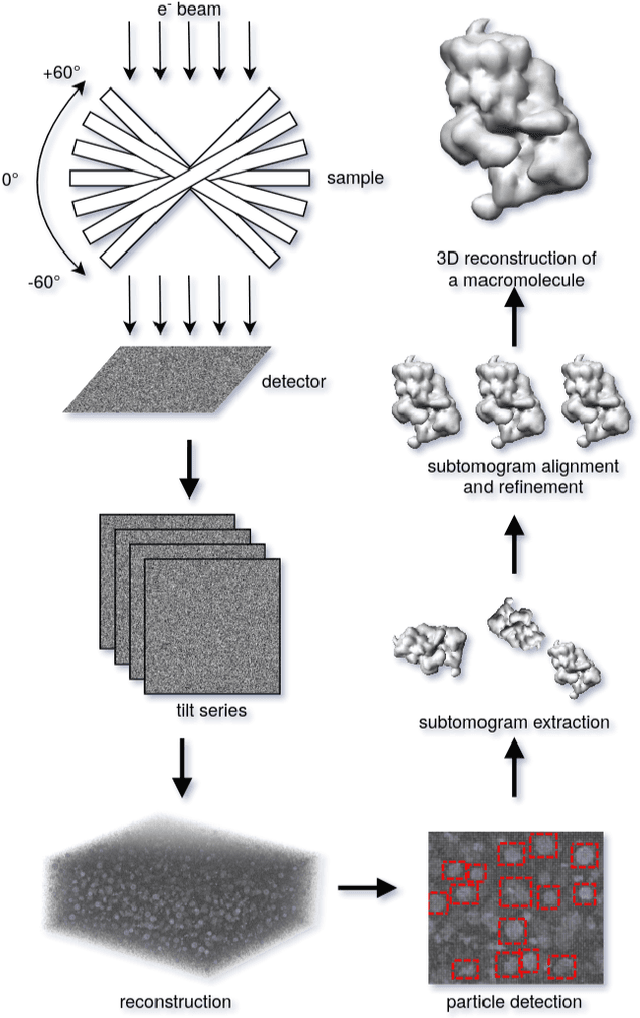
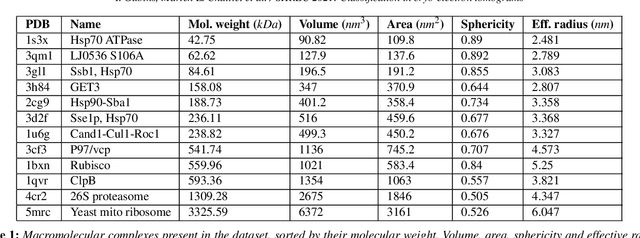
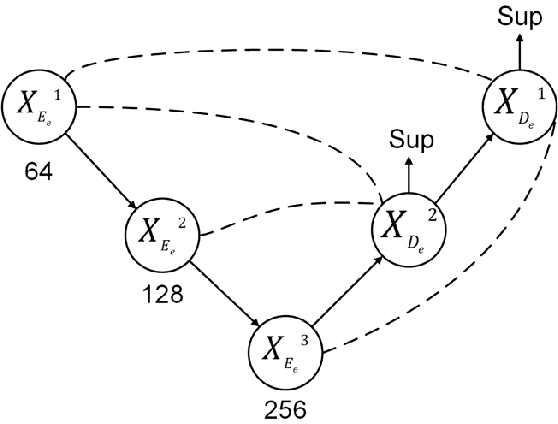
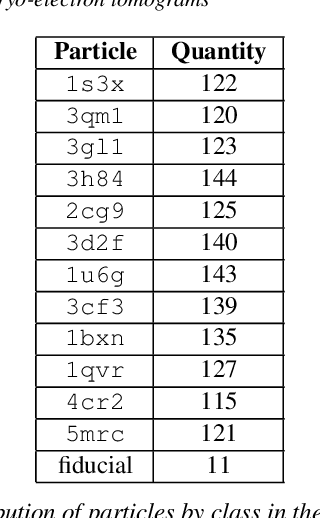
Cryo-electron tomography (cryo-ET) is an imaging technique that allows three-dimensional visualization of macro-molecular assemblies under near-native conditions. Cryo-ET comes with a number of challenges, mainly low signal-to-noise and inability to obtain images from all angles. Computational methods are key to analyze cryo-electron tomograms. To promote innovation in computational methods, we generate a novel simulated dataset to benchmark different methods of localization and classification of biological macromolecules in tomograms. Our publicly available dataset contains ten tomographic reconstructions of simulated cell-like volumes. Each volume contains twelve different types of complexes, varying in size, function and structure. In this paper, we have evaluated seven different methods of finding and classifying proteins. Seven research groups present results obtained with learning-based methods and trained on the simulated dataset, as well as a baseline template matching (TM), a traditional method widely used in cryo-ET research. We show that learning-based approaches can achieve notably better localization and classification performance than TM. We also experimentally confirm that there is a negative relationship between particle size and performance for all methods.