 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePF-DAformer: Proximal Femur Segmentation via Domain Adaptive Transformer for Dual-Center QCT
Oct 30, 2025Quantitative computed tomography (QCT) plays a crucial role in assessing bone strength and fracture risk by enabling volumetric analysis of bone density distribution in the proximal femur. However, deploying automated segmentation models in practice remains difficult because deep networks trained on one dataset often fail when applied to another. This failure stems from domain shift, where scanners, reconstruction settings, and patient demographics vary across institutions, leading to unstable predictions and unreliable quantitative metrics. Overcoming this barrier is essential for multi-center osteoporosis research and for ensuring that radiomics and structural finite element analysis results remain reproducible across sites. In this work, we developed a domain-adaptive transformer segmentation framework tailored for multi-institutional QCT. Our model is trained and validated on one of the largest hip fracture related research cohorts to date, comprising 1,024 QCT images scans from Tulane University and 384 scans from Rochester, Minnesota for proximal femur segmentation. To address domain shift, we integrate two complementary strategies within a 3D TransUNet backbone: adversarial alignment via Gradient Reversal Layer (GRL), which discourages the network from encoding site-specific cues, and statistical alignment via Maximum Mean Discrepancy (MMD), which explicitly reduces distributional mismatches between institutions. This dual mechanism balances invariance and fine-grained alignment, enabling scanner-agnostic feature learning while preserving anatomical detail.
UPMAD-Net: A Brain Tumor Segmentation Network with Uncertainty Guidance and Adaptive Multimodal Feature Fusion
May 06, 2025Background: Brain tumor segmentation has a significant impact on the diagnosis and treatment of brain tumors. Accurate brain tumor segmentation remains challenging due to their irregular shapes, vague boundaries, and high variability. Objective: We propose a brain tumor segmentation method that combines deep learning with prior knowledge derived from a region-growing algorithm. Methods: The proposed method utilizes a multi-scale feature fusion (MSFF) module and adaptive attention mechanisms (AAM) to extract multi-scale features and capture global contextual information. To enhance the model's robustness in low-confidence regions, the Monte Carlo Dropout (MC Dropout) strategy is employed for uncertainty estimation. Results: Extensive experiments demonstrate that the proposed method achieves superior performance on Brain Tumor Segmentation (BraTS) datasets, significantly outperforming various state-of-the-art methods. On the BraTS2021 dataset, the test Dice scores are 89.18% for Enhancing Tumor (ET) segmentation, 93.67% for Whole Tumor (WT) segmentation, and 91.23% for Tumor Core (TC) segmentation. On the BraTS2019 validation set, the validation Dice scores are 87.43%, 90.92%, and 90.40% for ET, WT, and TC segmentation, respectively. Ablation studies further confirmed the contribution of each module to segmentation accuracy, indicating that each component played a vital role in overall performance improvement. Conclusion: This study proposed a novel 3D brain tumor segmentation network based on the U-Net architecture. By incorporating the prior knowledge and employing the uncertainty estimation method, the robustness and performance were improved. The code for the proposed method is available at https://github.com/chenzhao2023/UPMAD_Net_BrainSeg.
Myocardial Region-guided Feature Aggregation Net for Automatic Coronary artery Segmentation and Stenosis Assessment using Coronary Computed Tomography Angiography
Apr 27, 2025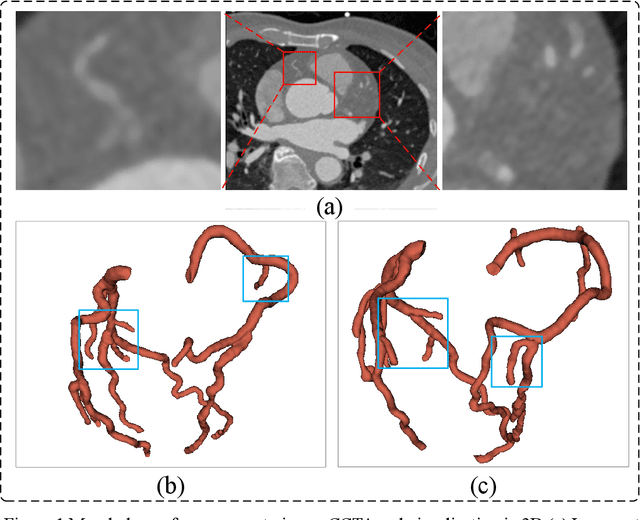
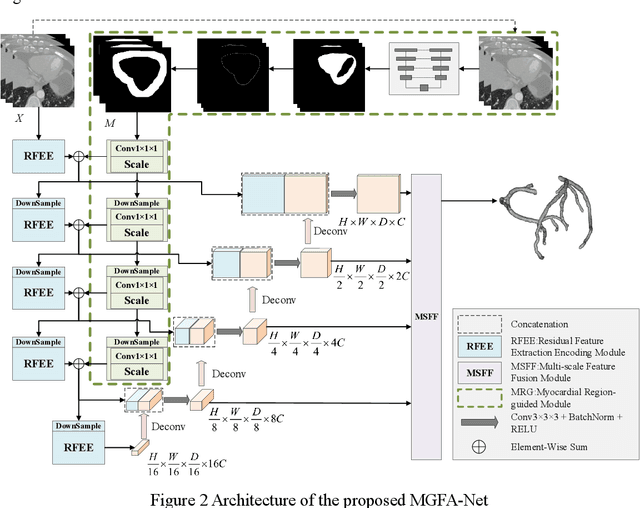
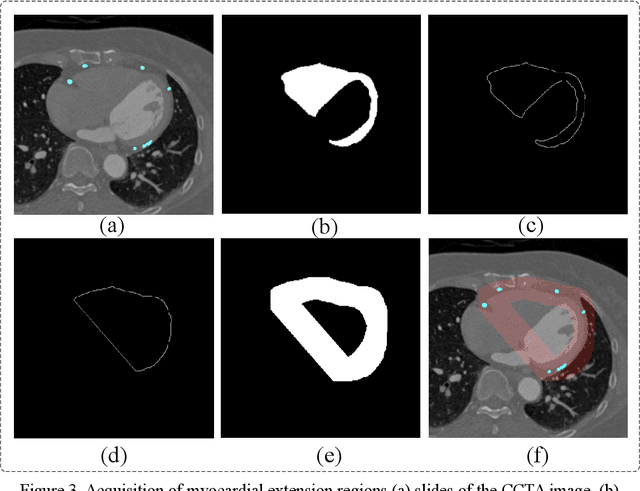
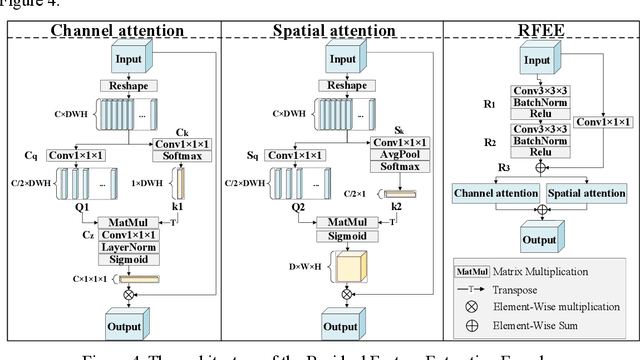
Coronary artery disease (CAD) remains a leading cause of mortality worldwide, requiring accurate segmentation and stenosis detection using Coronary Computed Tomography angiography (CCTA). Existing methods struggle with challenges such as low contrast, morphological variability and small vessel segmentation. To address these limitations, we propose the Myocardial Region-guided Feature Aggregation Net, a novel U-shaped dual-encoder architecture that integrates anatomical prior knowledge to enhance robustness in coronary artery segmentation. Our framework incorporates three key innovations: (1) a Myocardial Region-guided Module that directs attention to coronary regions via myocardial contour expansion and multi-scale feature fusion, (2) a Residual Feature Extraction Encoding Module that combines parallel spatial channel attention with residual blocks to enhance local-global feature discrimination, and (3) a Multi-scale Feature Fusion Module for adaptive aggregation of hierarchical vascular features. Additionally, Monte Carlo dropout f quantifies prediction uncertainty, supporting clinical interpretability. For stenosis detection, a morphology-based centerline extraction algorithm separates the vascular tree into anatomical branches, enabling cross-sectional area quantification and stenosis grading. The superiority of MGFA-Net was demonstrated by achieving an Dice score of 85.04%, an accuracy of 84.24%, an HD95 of 6.1294 mm, and an improvement of 5.46% in true positive rate for stenosis detection compared to3D U-Net. The integrated segmentation-to-stenosis pipeline provides automated, clinically interpretable CAD assessment, bridging deep learning with anatomical prior knowledge for precision medicine. Our code is publicly available at http://github.com/chenzhao2023/MGFA_CCTA
ICGM-FRAX: Iterative Cross Graph Matching for Hip Fracture Risk Assessment using Dual-energy X-ray Absorptiometry Images
Apr 21, 2025Hip fractures represent a major health concern, particularly among the elderly, often leading decreased mobility and increased mortality. Early and accurate detection of at risk individuals is crucial for effective intervention. In this study, we propose Iterative Cross Graph Matching for Hip Fracture Risk Assessment (ICGM-FRAX), a novel approach for predicting hip fractures using Dual-energy X-ray Absorptiometry (DXA) images. ICGM-FRAX involves iteratively comparing a test (subject) graph with multiple template graphs representing the characteristics of hip fracture subjects to assess the similarity and accurately to predict hip fracture risk. These graphs are obtained as follows. The DXA images are separated into multiple regions of interest (RoIs), such as the femoral head, shaft, and lesser trochanter. Radiomic features are then calculated for each RoI, with the central coordinates used as nodes in a graph. The connectivity between nodes is established according to the Euclidean distance between these coordinates. This process transforms each DXA image into a graph, where each node represents a RoI, and edges derived by the centroids of RoIs capture the spatial relationships between them. If the test graph closely matches a set of template graphs representing subjects with incident hip fractures, it is classified as indicating high hip fracture risk. We evaluated our method using 547 subjects from the UK Biobank dataset, and experimental results show that ICGM-FRAX achieved a sensitivity of 0.9869, demonstrating high accuracy in predicting hip fractures.
FedDA-TSformer: Federated Domain Adaptation with Vision TimeSformer for Left Ventricle Segmentation on Gated Myocardial Perfusion SPECT Image
Feb 23, 2025Background and Purpose: Functional assessment of the left ventricle using gated myocardial perfusion (MPS) single-photon emission computed tomography relies on the precise extraction of the left ventricular contours while simultaneously ensuring the security of patient data. Methods: In this paper, we introduce the integration of Federated Domain Adaptation with TimeSformer, named 'FedDA-TSformer' for left ventricle segmentation using MPS. FedDA-TSformer captures spatial and temporal features in gated MPS images, leveraging spatial attention, temporal attention, and federated learning for improved domain adaptation while ensuring patient data security. In detail, we employed Divide-Space-Time-Attention mechanism to extract spatio-temporal correlations from the multi-centered MPS datasets, ensuring that predictions are spatio-temporally consistent. To achieve domain adaptation, we align the model output on MPS from three different centers using local maximum mean discrepancy (LMMD) loss. This approach effectively addresses the dual requirements of federated learning and domain adaptation, enhancing the model's performance during training with multi-site datasets while ensuring the protection of data from different hospitals. Results: Our FedDA-TSformer was trained and evaluated using MPS datasets collected from three hospitals, comprising a total of 150 subjects. Each subject's cardiac cycle was divided into eight gates. The model achieved Dice Similarity Coefficients (DSC) of 0.842 and 0.907 for left ventricular (LV) endocardium and epicardium segmentation, respectively. Conclusion: Our proposed FedDA-TSformer model addresses the challenge of multi-center generalization, ensures patient data privacy protection, and demonstrates effectiveness in left ventricular (LV) segmentation.
MsMorph: An Unsupervised pyramid learning network for brain image registration
Oct 23, 2024In the field of medical image analysis, image registration is a crucial technique. Despite the numerous registration models that have been proposed, existing methods still fall short in terms of accuracy and interpretability. In this paper, we present MsMorph, a deep learning-based image registration framework aimed at mimicking the manual process of registering image pairs to achieve more similar deformations, where the registered image pairs exhibit consistency or similarity in features. By extracting the feature differences between image pairs across various as-pects using gradients, the framework decodes semantic information at different scales and continuously compen-sates for the predicted deformation field, driving the optimization of parameters to significantly improve registration accuracy. The proposed method simulates the manual approach to registration, focusing on different regions of the image pairs and their neighborhoods to predict the deformation field between the two images, which provides strong interpretability. We compared several existing registration methods on two public brain MRI datasets, including LPBA and Mindboggle. The experimental results show that our method consistently outperforms state of the art in terms of metrics such as Dice score, Hausdorff distance, average symmetric surface distance, and non-Jacobian. The source code is publicly available at https://github.com/GaodengFan/MsMorph
SGUQ: Staged Graph Convolution Neural Network for Alzheimer's Disease Diagnosis using Multi-Omics Data
Oct 14, 2024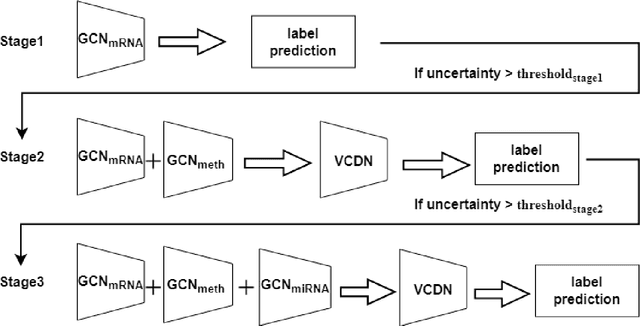

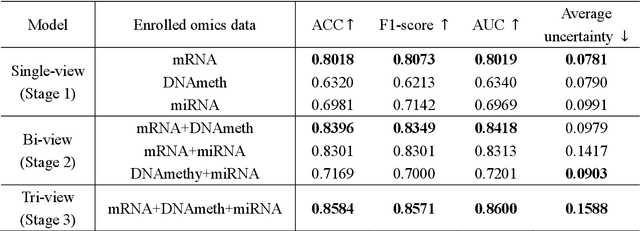
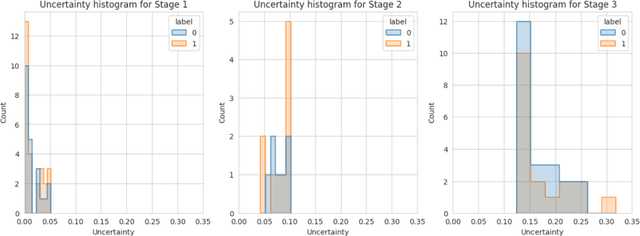
Alzheimer's disease (AD) is a chronic neurodegenerative disorder and the leading cause of dementia, significantly impacting cost, mortality, and burden worldwide. The advent of high-throughput omics technologies, such as genomics, transcriptomics, proteomics, and epigenomics, has revolutionized the molecular understanding of AD. Conventional AI approaches typically require the completion of all omics data at the outset to achieve optimal AD diagnosis, which are inefficient and may be unnecessary. To reduce the clinical cost and improve the accuracy of AD diagnosis using multi-omics data, we propose a novel staged graph convolutional network with uncertainty quantification (SGUQ). SGUQ begins with mRNA and progressively incorporates DNA methylation and miRNA data only when necessary, reducing overall costs and exposure to harmful tests. Experimental results indicate that 46.23% of the samples can be reliably predicted using only single-modal omics data (mRNA), while an additional 16.04% of the samples can achieve reliable predictions when combining two omics data types (mRNA + DNA methylation). In addition, the proposed staged SGUQ achieved an accuracy of 0.858 on ROSMAP dataset, which outperformed existing methods significantly. The proposed SGUQ can not only be applied to AD diagnosis using multi-omics data but also has the potential for clinical decision-making using multi-viewed data. Our implementation is publicly available at https://github.com/chenzhao2023/multiomicsuncertainty.
A Staged Approach using Machine Learning and Uncertainty Quantification to Predict the Risk of Hip Fracture
May 30, 2024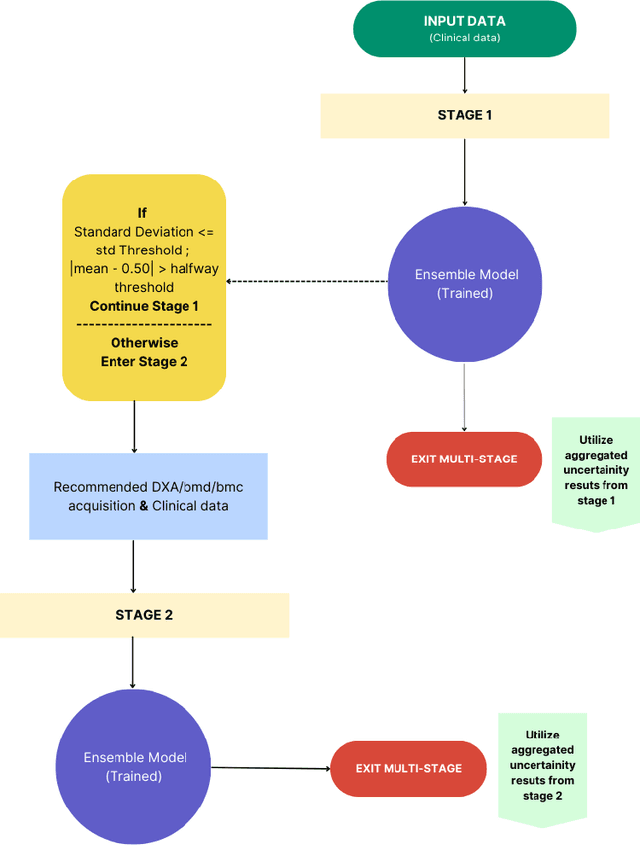
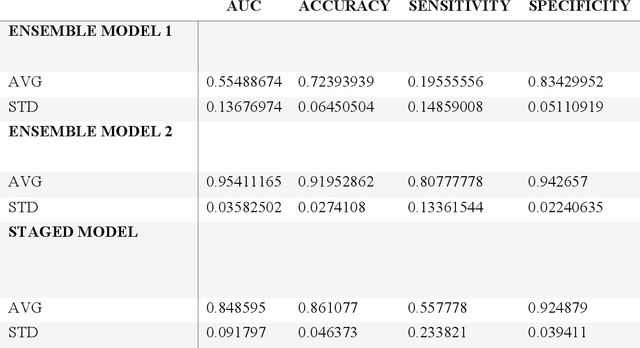
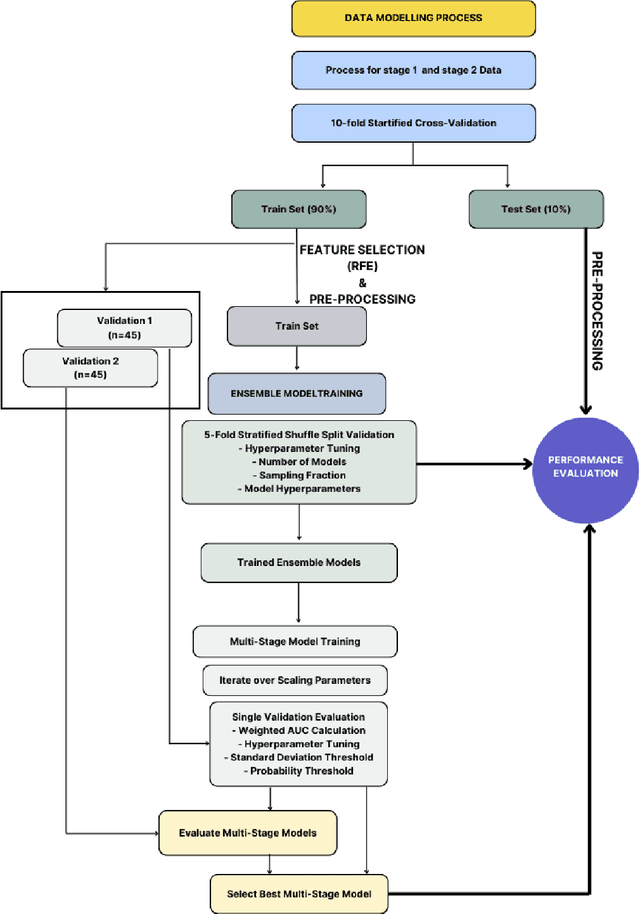
Despite advancements in medical care, hip fractures impose a significant burden on individuals and healthcare systems. This paper focuses on the prediction of hip fracture risk in older and middle-aged adults, where falls and compromised bone quality are predominant factors. We propose a novel staged model that combines advanced imaging and clinical data to improve predictive performance. By using CNNs to extract features from hip DXA images, along with clinical variables, shape measurements, and texture features, our method provides a comprehensive framework for assessing fracture risk. A staged machine learning-based model was developed using two ensemble models: Ensemble 1 (clinical variables only) and Ensemble 2 (clinical variables and DXA imaging features). This staged approach used uncertainty quantification from Ensemble 1 to decide if DXA features are necessary for further prediction. Ensemble 2 exhibited the highest performance, achieving an AUC of 0.9541, an accuracy of 0.9195, a sensitivity of 0.8078, and a specificity of 0.9427. The staged model also performed well, with an AUC of 0.8486, an accuracy of 0.8611, a sensitivity of 0.5578, and a specificity of 0.9249, outperforming Ensemble 1, which had an AUC of 0.5549, an accuracy of 0.7239, a sensitivity of 0.1956, and a specificity of 0.8343. Furthermore, the staged model suggested that 54.49% of patients did not require DXA scanning. It effectively balanced accuracy and specificity, offering a robust solution when DXA data acquisition is not always feasible. Statistical tests confirmed significant differences between the models, highlighting the advantages of the advanced modeling strategies. Our staged approach could identify individuals at risk with a high accuracy but reduce the unnecessary DXA scanning. It has great promise to guide interventions to prevent hip fractures with reduced cost and radiation.
Multi-graph Graph Matching for Coronary Artery Semantic Labeling
Feb 24, 2024Coronary artery disease (CAD) stands as the leading cause of death worldwide, and invasive coronary angiography (ICA) remains the gold standard for assessing vascular anatomical information. However, deep learning-based methods encounter challenges in generating semantic labels for arterial segments, primarily due to the morphological similarity between arterial branches. To address this challenge, we model the vascular tree as a graph and propose a multi-graph graph matching (MGM) algorithm for coronary artery semantic labeling. The MGM algorithm assesses the similarity between arterials in multiple vascular tree graphs, taking into account the cycle consistency between each pair of graphs. This ensures that unannotated arterial segments are appropriately labeled by matching them with annotated segments. Through the incorporation of anatomical graph structure, radiomics features, and semantic mapping, the proposed MGM model achieves an impressive accuracy of 0.9471 for coronary artery semantic labeling. This approach presents a novel tool for coronary artery analysis using ICA videos, offering valuable insights into vascular health and pathology.
Point cloud-based registration and image fusion between cardiac SPECT MPI and CTA
Feb 10, 2024A method was proposed for the point cloud-based registration and image fusion between cardiac single photon emission computed tomography (SPECT) myocardial perfusion images (MPI) and cardiac computed tomography angiograms (CTA). Firstly, the left ventricle (LV) epicardial regions (LVERs) in SPECT and CTA images were segmented by using different U-Net neural networks trained to generate the point clouds of the LV epicardial contours (LVECs). Secondly, according to the characteristics of cardiac anatomy, the special points of anterior and posterior interventricular grooves (APIGs) were manually marked in both SPECT and CTA image volumes. Thirdly, we developed an in-house program for coarsely registering the special points of APIGs to ensure a correct cardiac orientation alignment between SPECT and CTA images. Fourthly, we employed ICP, SICP or CPD algorithm to achieve a fine registration for the point clouds (together with the special points of APIGs) of the LV epicardial surfaces (LVERs) in SPECT and CTA images. Finally, the image fusion between SPECT and CTA was realized after the fine registration. The experimental results showed that the cardiac orientation was aligned well and the mean distance error of the optimal registration method (CPD with affine transform) was consistently less than 3 mm. The proposed method could effectively fuse the structures from cardiac CTA and SPECT functional images, and demonstrated a potential in assisting in accurate diagnosis of cardiac diseases by combining complementary advantages of the two imaging modalities.