 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeICGM-FRAX: Iterative Cross Graph Matching for Hip Fracture Risk Assessment using Dual-energy X-ray Absorptiometry Images
Apr 21, 2025Hip fractures represent a major health concern, particularly among the elderly, often leading decreased mobility and increased mortality. Early and accurate detection of at risk individuals is crucial for effective intervention. In this study, we propose Iterative Cross Graph Matching for Hip Fracture Risk Assessment (ICGM-FRAX), a novel approach for predicting hip fractures using Dual-energy X-ray Absorptiometry (DXA) images. ICGM-FRAX involves iteratively comparing a test (subject) graph with multiple template graphs representing the characteristics of hip fracture subjects to assess the similarity and accurately to predict hip fracture risk. These graphs are obtained as follows. The DXA images are separated into multiple regions of interest (RoIs), such as the femoral head, shaft, and lesser trochanter. Radiomic features are then calculated for each RoI, with the central coordinates used as nodes in a graph. The connectivity between nodes is established according to the Euclidean distance between these coordinates. This process transforms each DXA image into a graph, where each node represents a RoI, and edges derived by the centroids of RoIs capture the spatial relationships between them. If the test graph closely matches a set of template graphs representing subjects with incident hip fractures, it is classified as indicating high hip fracture risk. We evaluated our method using 547 subjects from the UK Biobank dataset, and experimental results show that ICGM-FRAX achieved a sensitivity of 0.9869, demonstrating high accuracy in predicting hip fractures.
A Staged Approach using Machine Learning and Uncertainty Quantification to Predict the Risk of Hip Fracture
May 30, 2024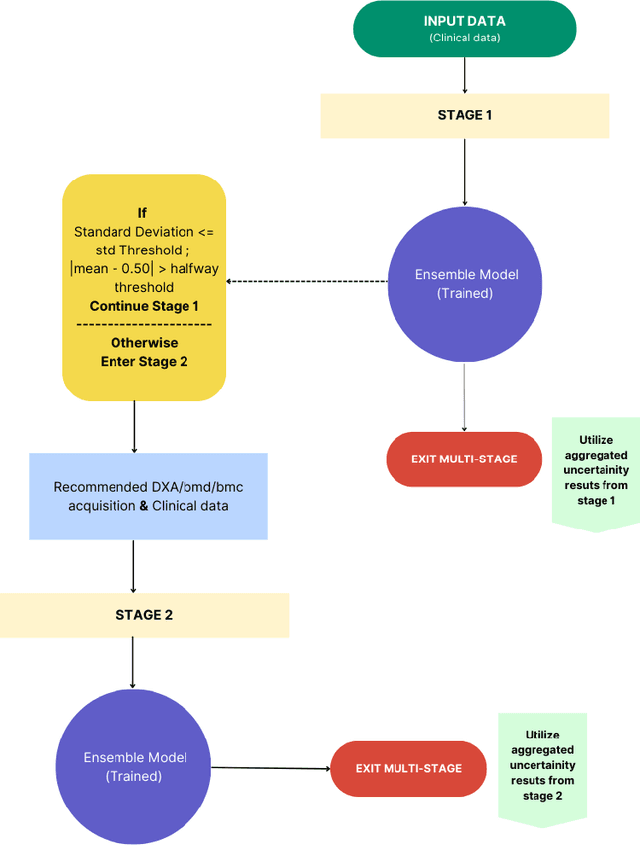
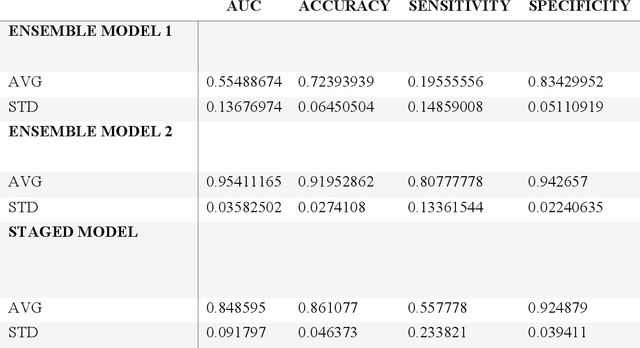
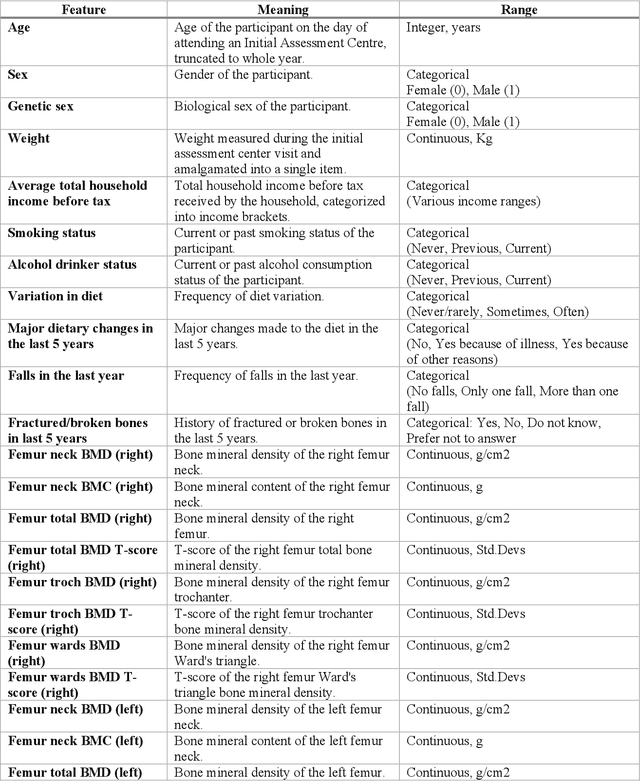
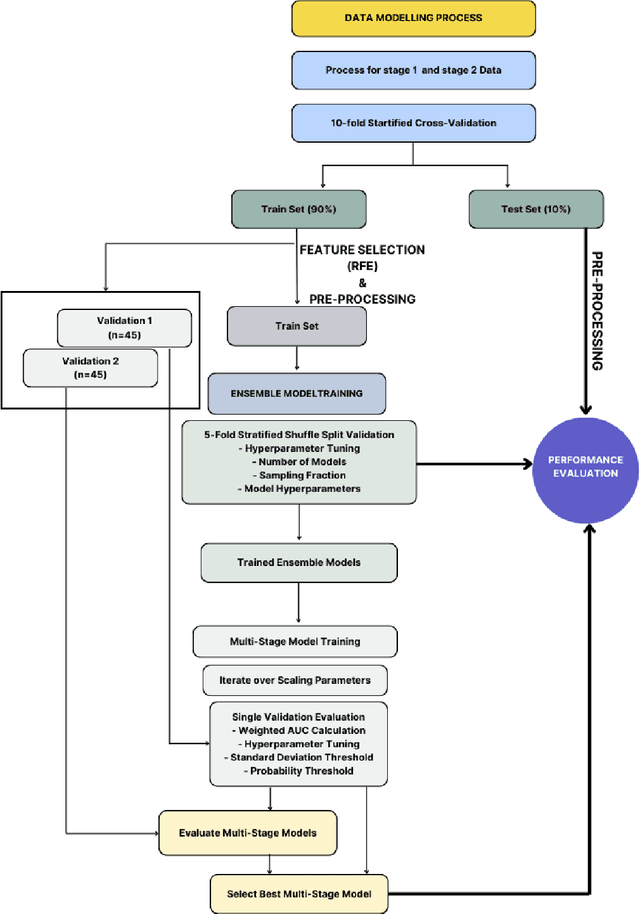
Despite advancements in medical care, hip fractures impose a significant burden on individuals and healthcare systems. This paper focuses on the prediction of hip fracture risk in older and middle-aged adults, where falls and compromised bone quality are predominant factors. We propose a novel staged model that combines advanced imaging and clinical data to improve predictive performance. By using CNNs to extract features from hip DXA images, along with clinical variables, shape measurements, and texture features, our method provides a comprehensive framework for assessing fracture risk. A staged machine learning-based model was developed using two ensemble models: Ensemble 1 (clinical variables only) and Ensemble 2 (clinical variables and DXA imaging features). This staged approach used uncertainty quantification from Ensemble 1 to decide if DXA features are necessary for further prediction. Ensemble 2 exhibited the highest performance, achieving an AUC of 0.9541, an accuracy of 0.9195, a sensitivity of 0.8078, and a specificity of 0.9427. The staged model also performed well, with an AUC of 0.8486, an accuracy of 0.8611, a sensitivity of 0.5578, and a specificity of 0.9249, outperforming Ensemble 1, which had an AUC of 0.5549, an accuracy of 0.7239, a sensitivity of 0.1956, and a specificity of 0.8343. Furthermore, the staged model suggested that 54.49% of patients did not require DXA scanning. It effectively balanced accuracy and specificity, offering a robust solution when DXA data acquisition is not always feasible. Statistical tests confirmed significant differences between the models, highlighting the advantages of the advanced modeling strategies. Our staged approach could identify individuals at risk with a high accuracy but reduce the unnecessary DXA scanning. It has great promise to guide interventions to prevent hip fractures with reduced cost and radiation.
Multi-View Variational Autoencoder for Missing Value Imputation in Untargeted Metabolomics
Oct 12, 2023


Background: Missing data is a common challenge in mass spectrometry-based metabolomics, which can lead to biased and incomplete analyses. The integration of whole-genome sequencing (WGS) data with metabolomics data has emerged as a promising approach to enhance the accuracy of data imputation in metabolomics studies. Method: In this study, we propose a novel method that leverages the information from WGS data and reference metabolites to impute unknown metabolites. Our approach utilizes a multi-view variational autoencoder to jointly model the burden score, polygenetic risk score (PGS), and linkage disequilibrium (LD) pruned single nucleotide polymorphisms (SNPs) for feature extraction and missing metabolomics data imputation. By learning the latent representations of both omics data, our method can effectively impute missing metabolomics values based on genomic information. Results: We evaluate the performance of our method on empirical metabolomics datasets with missing values and demonstrate its superiority compared to conventional imputation techniques. Using 35 template metabolites derived burden scores, PGS and LD-pruned SNPs, the proposed methods achieved r2-scores > 0.01 for 71.55% of metabolites. Conclusion: The integration of WGS data in metabolomics imputation not only improves data completeness but also enhances downstream analyses, paving the way for more comprehensive and accurate investigations of metabolic pathways and disease associations. Our findings offer valuable insights into the potential benefits of utilizing WGS data for metabolomics data imputation and underscore the importance of leveraging multi-modal data integration in precision medicine research.
A new method of modeling the multi-stage decision-making process of CRT using machine learning with uncertainty quantification
Sep 19, 2023
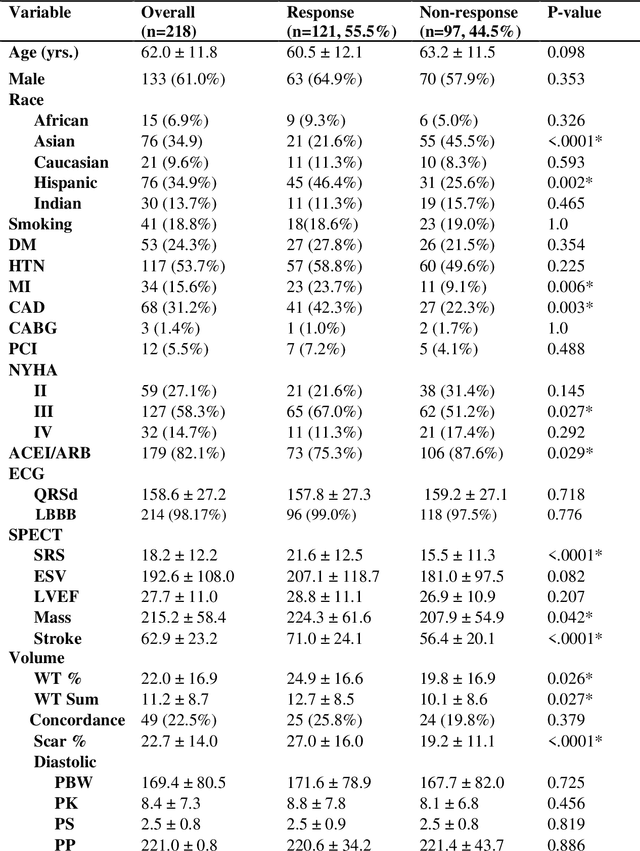
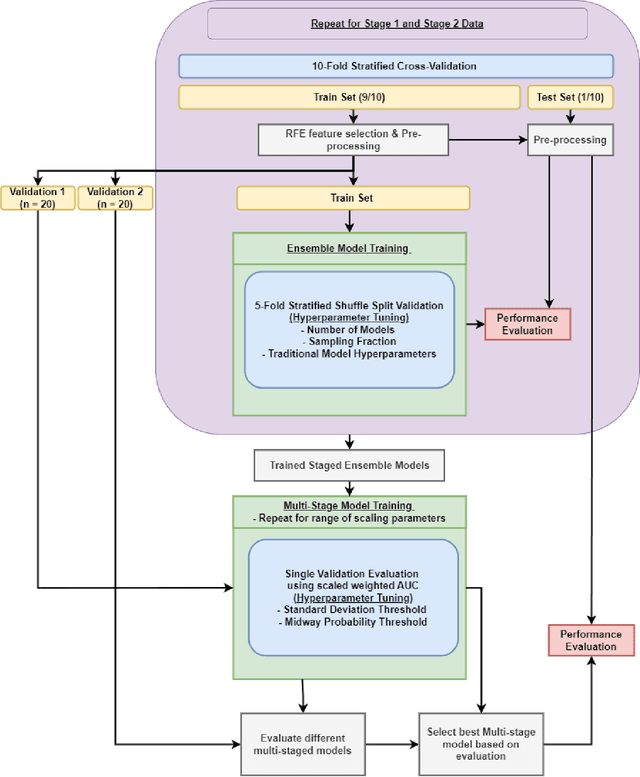
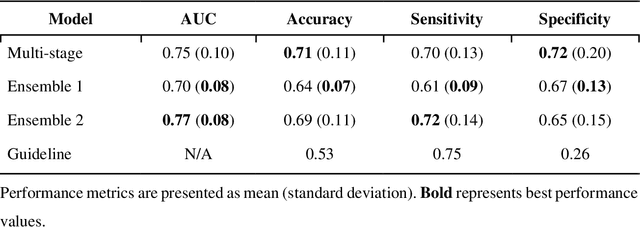
Aims. The purpose of this study is to create a multi-stage machine learning model to predict cardiac resynchronization therapy (CRT) response for heart failure (HF) patients. This model exploits uncertainty quantification to recommend additional collection of single-photon emission computed tomography myocardial perfusion imaging (SPECT MPI) variables if baseline clinical variables and features from electrocardiogram (ECG) are not sufficient. Methods. 218 patients who underwent rest-gated SPECT MPI were enrolled in this study. CRT response was defined as an increase in left ventricular ejection fraction (LVEF) > 5% at a 6 month follow-up. A multi-stage ML model was created by combining two ensemble models. Results. The response rate for CRT was 55.5% (n = 121) with overall male gender 61.0% (n = 133), an average age of 62.0, and LVEF of 27.7. The multi-stage model performed similarly to Ensemble 2 (which utilized the additional SPECT data) with AUC of 0.75 vs. 0.77, accuracy of 0.71 vs. 0.69, sensitivity of 0.70 vs. 0.72, and specificity 0.72 vs. 0.65, respectively. However, the multi-stage model only required SPECT MPI data for 52.7% of the patients across all folds. Conclusions. By using rule-based logic stemming from uncertainty quantification, the multi-stage model was able to reduce the need for additional SPECT MPI data acquisition without sacrificing performance.
CLCLSA: Cross-omics Linked embedding with Contrastive Learning and Self Attention for multi-omics integration with incomplete multi-omics data
Apr 12, 2023



Integration of heterogeneous and high-dimensional multi-omics data is becoming increasingly important in understanding genetic data. Each omics technique only provides a limited view of the underlying biological process and integrating heterogeneous omics layers simultaneously would lead to a more comprehensive and detailed understanding of diseases and phenotypes. However, one obstacle faced when performing multi-omics data integration is the existence of unpaired multi-omics data due to instrument sensitivity and cost. Studies may fail if certain aspects of the subjects are missing or incomplete. In this paper, we propose a deep learning method for multi-omics integration with incomplete data by Cross-omics Linked unified embedding with Contrastive Learning and Self Attention (CLCLSA). Utilizing complete multi-omics data as supervision, the model employs cross-omics autoencoders to learn the feature representation across different types of biological data. The multi-omics contrastive learning, which is used to maximize the mutual information between different types of omics, is employed before latent feature concatenation. In addition, the feature-level self-attention and omics-level self-attention are employed to dynamically identify the most informative features for multi-omics data integration. Extensive experiments were conducted on four public multi-omics datasets. The experimental results indicated that the proposed CLCLSA outperformed the state-of-the-art approaches for multi-omics data classification using incomplete multi-omics data.
A new method using machine learning to integrate ECG and gated SPECT MPI for Cardiac Resynchronization Therapy Decision Support on behalf of the VISION-CRT
Nov 06, 2022Cardiac resynchronization therapy (CRT) has been established as an important therapy for heart failure. Mechanical dyssynchrony has the potential to predict responders to CRT. The aim of this study was to report the development and the validation of machine learning (ML) models which integrates ECG, gated SPECT MPI (GMPS) and clinical variables to predict patients' response to CRT. This analysis included 153 patients who met criteria for CRT from a prospective cohort study. The variables were used to modeling predictive methods for CRT. Patients were classified as responders for an increase of LVEF>=5% at follow-up. In a second analysis, patients were classified super-responders for increase of LVEF>=15%. For ML, variable selection was applied, and Prediction Analysis of Microarrays (PAM) approach was used for response modeling while Naive Bayes (NB) was used for super-response. They were compared to models obtained with guideline variables. PAM had AUC of 0.80 against 0.71 of logistic regression with guideline variables (p = 0.47). The sensitivity (0.86) and specificity (0.75) were better than for guideline alone, sensitivity (0.72) and specificity (0.22). Neural network with guideline variables outperformed NB (AUC = 0.87 vs 0.86; p = 0.88). Its sensitivity and specificity (1.0 and 0.75, respectively) was better than guideline alone (0.40 and 0.06, respectively). Compared to guideline criteria, ML methods trended towards improved CRT response and super-response prediction. GMPS had a central role in the acquisition of most parameters. Further studies are needed to validate the models.
Multi-view information fusion using multi-view variational autoencoders to predict proximal femoral strength
Oct 03, 2022
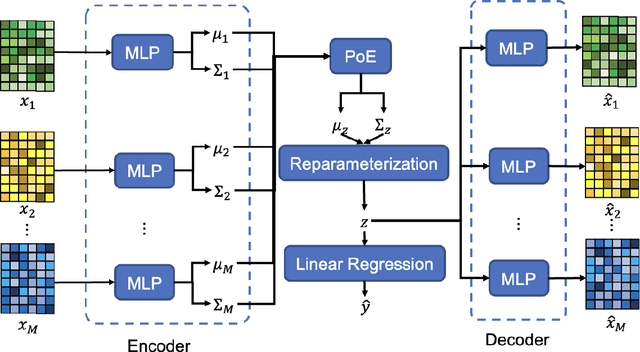
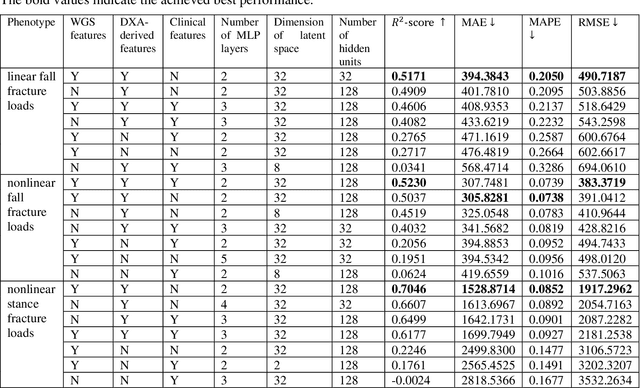
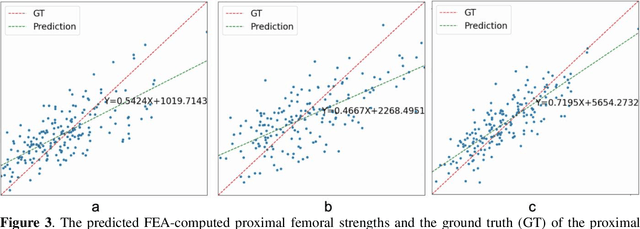
Background and aim: Hip fracture can be devastating. The proximal femoral strength can be computed by subject-specific finite element (FE) analysis (FEA) using quantitative CT images. The aim of this paper is to design a deep learning-based model for hip fracture prediction with multi-view information fusion. Method: We developed a multi-view variational autoencoder (MMVAE) for feature representation learning and designed the product of expert model (PoE) for multi-view information fusion.We performed genome-wide association studies (GWAS) to select the most relevant genetic features with proximal femoral strengths and integrated genetic features with DXA-derived imaging features and clinical variables for proximal femoral strength prediction. Results: The designed model achieved the mean absolute percentage error of 0.2050,0.0739 and 0.0852 for linear fall, nonlinear fall and nonlinear stance fracture load prediction, respectively. For linear fall and nonlinear stance fracture load prediction, integrating genetic and DXA-derived imaging features were beneficial; while for nonlinear fall fracture load prediction, integrating genetic features, DXA-derived imaging features as well as clinical variables, the model achieved the best performance. Conclusion: The proposed model is capable of predicting proximal femoral strengths using genetic features, DXA-derived imaging features as well as clinical variables. Compared to performing FEA using QCT images to calculate proximal femoral strengths, the presented method is time-efficient and cost effective, and radiation dosage is limited. From the technique perspective, the final models can be applied to other multi-view information integration tasks.
Hip Fracture Prediction using the First Principal Component Derived from FEA-Computed Fracture Loads
Oct 03, 2022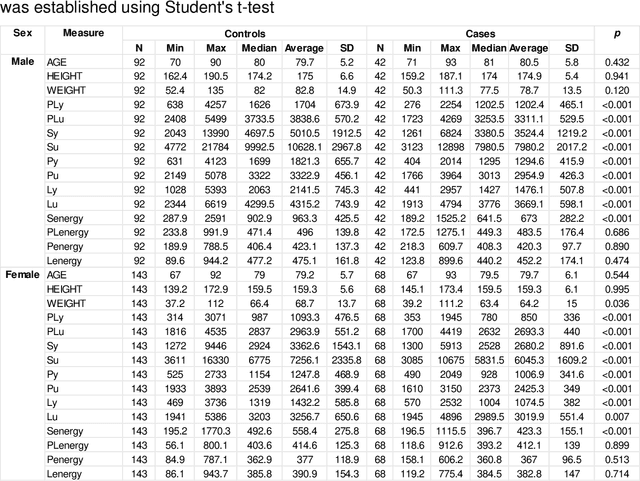
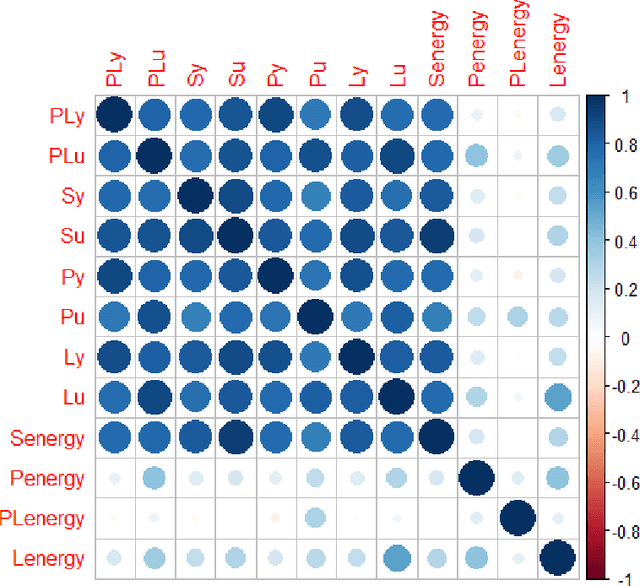
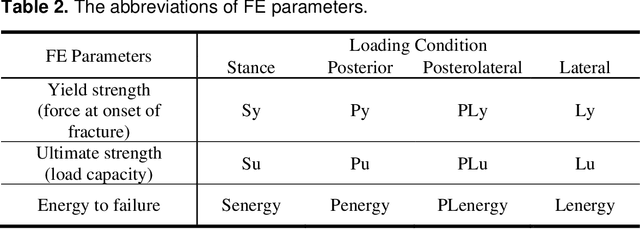
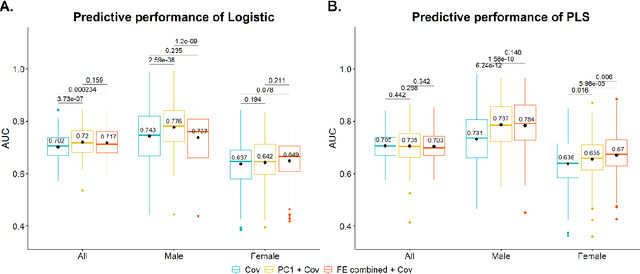
Hip fracture risk assessment is an important but challenging task. Quantitative CT-based patient specific finite element analysis (FEA) computes the force (fracture load) to break the proximal femur in a particular loading condition. It provides different structural information about the proximal femur that can influence a subject overall fracture risk. To obtain a more robust measure of fracture risk, we used principal component analysis (PCA) to develop a global FEA computed fracture risk index that incorporates the FEA-computed yield and ultimate failure loads and energies to failure in four loading conditions (single-limb stance and impact from a fall onto the posterior, posterolateral, and lateral aspects of the greater trochanter) of 110 hip fracture subjects and 235 age and sex matched control subjects from the AGES-Reykjavik study. We found that the first PC (PC1) of the FE parameters was the only significant predictor of hip fracture. Using a logistic regression model, we determined if prediction performance for hip fracture using PC1 differed from that using FE parameters combined by stratified random resampling with respect to hip fracture status. The results showed that the average of the area under the receive operating characteristic curve (AUC) using PC1 was always higher than that using all FE parameters combined in the male subjects. The AUC of PC1 and AUC of the FE parameters combined were not significantly different than that in the female subjects or in all subjects