 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBeyond Attention Heatmaps: How to Get Better Explanations for Multiple Instance Learning Models in Histopathology
Mar 09, 2026Multiple instance learning (MIL) has enabled substantial progress in computational histopathology, where a large amount of patches from gigapixel whole slide images are aggregated into slide-level predictions. Heatmaps are widely used to validate MIL models and to discover tissue biomarkers. Yet, the validity of these heatmaps has barely been investigated. In this work, we introduce a general framework for evaluating the quality of MIL heatmaps without requiring additional labels. We conduct a large-scale benchmark experiment to assess six explanation methods across histopathology task types (classification, regression, survival), MIL model architectures (Attention-, Transformer-, Mamba-based), and patch encoder backbones (UNI2, Virchow2). Our results show that explanation quality mostly depends on MIL model architecture and task type, with perturbation ("Single"), layer-wise relevance propagation (LRP), and integrated gradients (IG) consistently outperforming attention-based and gradient-based saliency heatmaps, which often fail to reflect model decision mechanisms. We further demonstrate the advanced capabilities of the best-performing explanation methods: (i) We provide a proof-of-concept that MIL heatmaps of a bulk gene expression prediction model can be correlated with spatial transcriptomics for biological validation, and (ii) showcase the discovery of distinct model strategies for predicting human papillomavirus (HPV) infection from head and neck cancer slides. Our work highlights the importance of validating MIL heatmaps and establishes that improved explainability can enable more reliable model validation and yield biological insights, making a case for a broader adoption of explainable AI in digital pathology. Our code is provided in a public GitHub repository: https://github.com/bifold-pathomics/xMIL/tree/xmil-journal
Atlas 2 - Foundation models for clinical deployment
Jan 08, 2026Pathology foundation models substantially advanced the possibilities in computational pathology -- yet tradeoffs in terms of performance, robustness, and computational requirements remained, which limited their clinical deployment. In this report, we present Atlas 2, Atlas 2-B, and Atlas 2-S, three pathology vision foundation models which bridge these shortcomings by showing state-of-the-art performance in prediction performance, robustness, and resource efficiency in a comprehensive evaluation across eighty public benchmarks. Our models were trained on the largest pathology foundation model dataset to date comprising 5.5 million histopathology whole slide images, collected from three medical institutions Charité - Universtätsmedizin Berlin, LMU Munich, and Mayo Clinic.
Towards Robust Foundation Models for Digital Pathology
Jul 22, 2025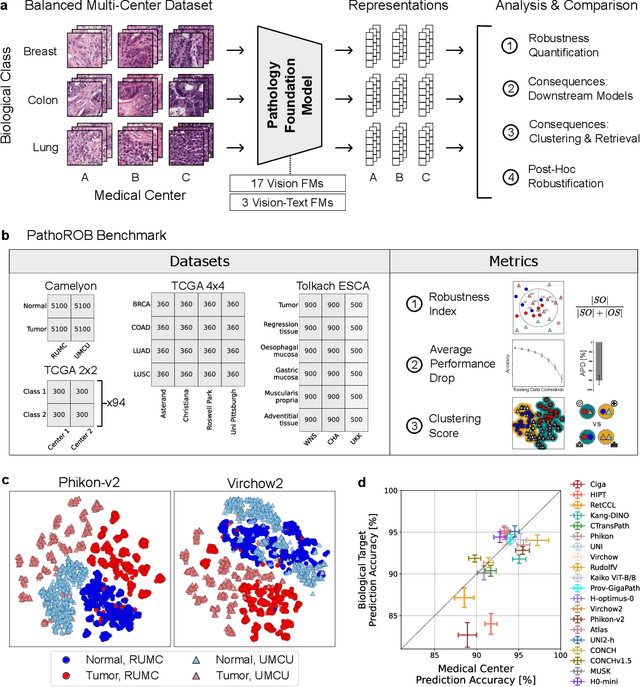
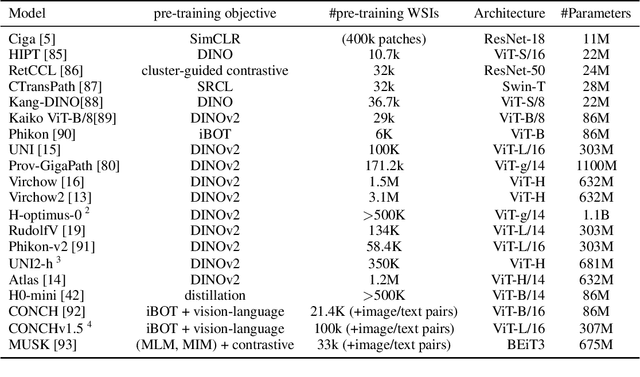
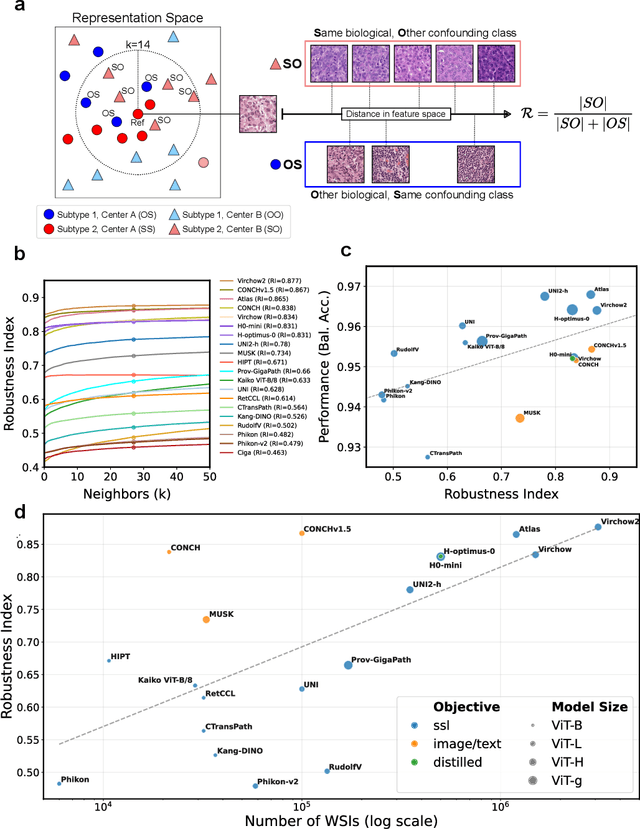

Biomedical Foundation Models (FMs) are rapidly transforming AI-enabled healthcare research and entering clinical validation. However, their susceptibility to learning non-biological technical features -- including variations in surgical/endoscopic techniques, laboratory procedures, and scanner hardware -- poses risks for clinical deployment. We present the first systematic investigation of pathology FM robustness to non-biological features. Our work (i) introduces measures to quantify FM robustness, (ii) demonstrates the consequences of limited robustness, and (iii) proposes a framework for FM robustification to mitigate these issues. Specifically, we developed PathoROB, a robustness benchmark with three novel metrics, including the robustness index, and four datasets covering 28 biological classes from 34 medical centers. Our experiments reveal robustness deficits across all 20 evaluated FMs, and substantial robustness differences between them. We found that non-robust FM representations can cause major diagnostic downstream errors and clinical blunders that prevent safe clinical adoption. Using more robust FMs and post-hoc robustification considerably reduced (but did not yet eliminate) the risk of such errors. This work establishes that robustness evaluation is essential for validating pathology FMs before clinical adoption and demonstrates that future FM development must integrate robustness as a core design principle. PathoROB provides a blueprint for assessing robustness across biomedical domains, guiding FM improvement efforts towards more robust, representative, and clinically deployable AI systems that prioritize biological information over technical artifacts.
Atlas: A Novel Pathology Foundation Model by Mayo Clinic, Charité, and Aignostics
Jan 10, 2025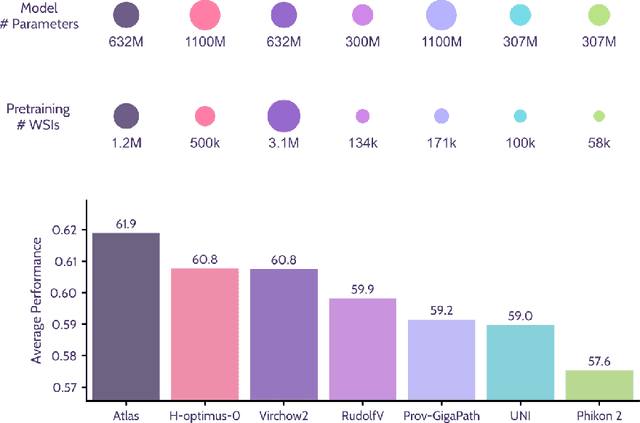
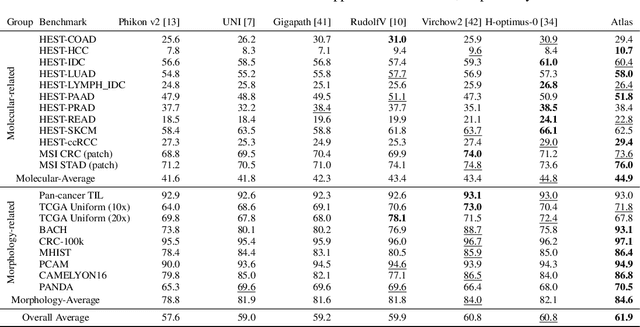
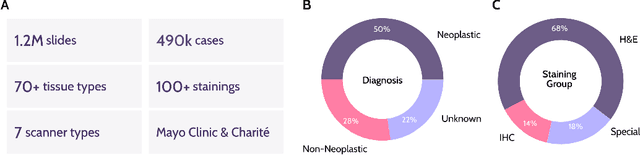
Recent advances in digital pathology have demonstrated the effectiveness of foundation models across diverse applications. In this report, we present Atlas, a novel vision foundation model based on the RudolfV approach. Our model was trained on a dataset comprising 1.2 million histopathology whole slide images, collected from two medical institutions: Mayo Clinic and Charit\'e - Universt\"atsmedizin Berlin. Comprehensive evaluations show that Atlas achieves state-of-the-art performance across twenty-one public benchmark datasets, even though it is neither the largest model by parameter count nor by training dataset size.
xCG: Explainable Cell Graphs for Survival Prediction in Non-Small Cell Lung Cancer
Nov 12, 2024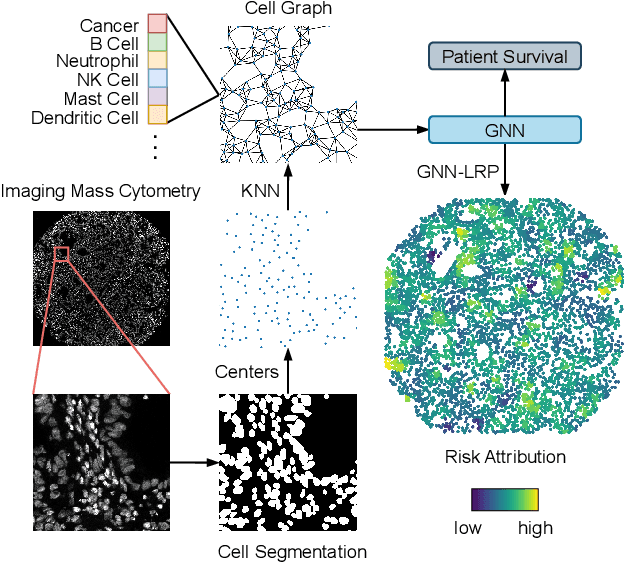
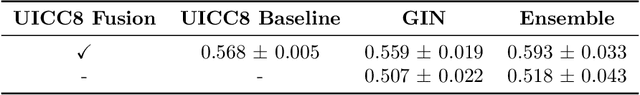
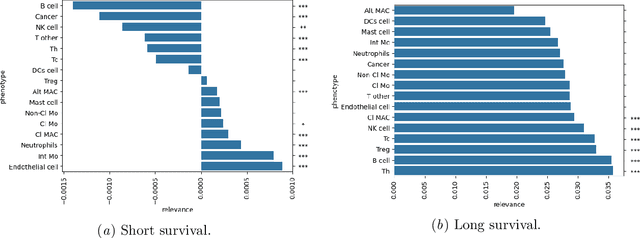
Understanding how deep learning models predict oncology patient risk can provide critical insights into disease progression, support clinical decision-making, and pave the way for trustworthy and data-driven precision medicine. Building on recent advances in the spatial modeling of the tumor microenvironment using graph neural networks, we present an explainable cell graph (xCG) approach for survival prediction. We validate our model on a public cohort of imaging mass cytometry (IMC) data for 416 cases of lung adenocarcinoma. We explain survival predictions in terms of known phenotypes on the cell level by computing risk attributions over cell graphs, for which we propose an efficient grid-based layer-wise relevance propagation (LRP) method. Our ablation studies highlight the importance of incorporating the cancer stage and model ensembling to improve the quality of risk estimates. Our xCG method, together with the IMC data, is made publicly available to support further research.
AI-based Anomaly Detection for Clinical-Grade Histopathological Diagnostics
Jun 21, 2024



While previous studies have demonstrated the potential of AI to diagnose diseases in imaging data, clinical implementation is still lagging behind. This is partly because AI models require training with large numbers of examples only available for common diseases. In clinical reality, however, only few diseases are common, whereas the majority of diseases are less frequent (long-tail distribution). Current AI models overlook or misclassify these diseases. We propose a deep anomaly detection approach that only requires training data from common diseases to detect also all less frequent diseases. We collected two large real-world datasets of gastrointestinal biopsies, which are prototypical of the problem. Herein, the ten most common findings account for approximately 90% of cases, whereas the remaining 10% contained 56 disease entities, including many cancers. 17 million histological images from 5,423 cases were used for training and evaluation. Without any specific training for the diseases, our best-performing model reliably detected a broad spectrum of infrequent ("anomalous") pathologies with 95.0% (stomach) and 91.0% (colon) AUROC and generalized across scanners and hospitals. By design, the proposed anomaly detection can be expected to detect any pathological alteration in the diagnostic tail of gastrointestinal biopsies, including rare primary or metastatic cancers. This study establishes the first effective clinical application of AI-based anomaly detection in histopathology that can flag anomalous cases, facilitate case prioritization, reduce missed diagnoses and enhance the general safety of AI models, thereby driving AI adoption and automation in routine diagnostics and beyond.
xMIL: Insightful Explanations for Multiple Instance Learning in Histopathology
Jun 06, 2024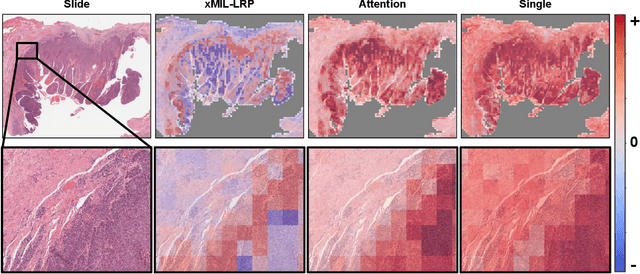
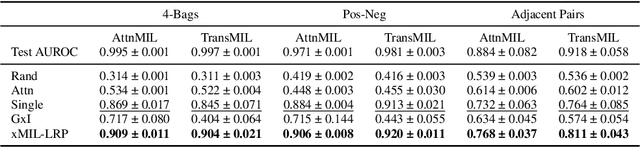
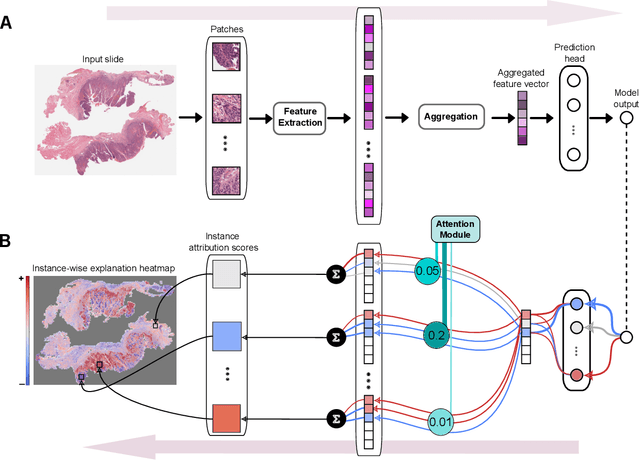
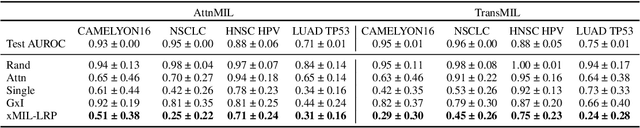
Multiple instance learning (MIL) is an effective and widely used approach for weakly supervised machine learning. In histopathology, MIL models have achieved remarkable success in tasks like tumor detection, biomarker prediction, and outcome prognostication. However, MIL explanation methods are still lagging behind, as they are limited to small bag sizes or disregard instance interactions. We revisit MIL through the lens of explainable AI (XAI) and introduce xMIL, a refined framework with more general assumptions. We demonstrate how to obtain improved MIL explanations using layer-wise relevance propagation (LRP) and conduct extensive evaluation experiments on three toy settings and four real-world histopathology datasets. Our approach consistently outperforms previous explanation attempts with particularly improved faithfulness scores on challenging biomarker prediction tasks. Finally, we showcase how xMIL explanations enable pathologists to extract insights from MIL models, representing a significant advance for knowledge discovery and model debugging in digital histopathology.
RudolfV: A Foundation Model by Pathologists for Pathologists
Jan 23, 2024



Histopathology plays a central role in clinical medicine and biomedical research. While artificial intelligence shows promising results on many pathological tasks, generalization and dealing with rare diseases, where training data is scarce, remains a challenge. Distilling knowledge from unlabeled data into a foundation model before learning from, potentially limited, labeled data provides a viable path to address these challenges. In this work, we extend the state of the art of foundation models for digital pathology whole slide images by semi-automated data curation and incorporating pathologist domain knowledge. Specifically, we combine computational and pathologist domain knowledge (1) to curate a diverse dataset of 103k slides corresponding to 750 million image patches covering data from different fixation, staining, and scanning protocols as well as data from different indications and labs across the EU and US, (2) for grouping semantically similar slides and tissue patches, and (3) to augment the input images during training. We evaluate the resulting model on a set of public and internal benchmarks and show that although our foundation model is trained with an order of magnitude less slides, it performs on par or better than competing models. We expect that scaling our approach to more data and larger models will further increase its performance and capacity to deal with increasingly complex real world tasks in diagnostics and biomedical research.
DiffInfinite: Large Mask-Image Synthesis via Parallel Random Patch Diffusion in Histopathology
Jun 23, 2023



We present DiffInfinite, a hierarchical diffusion model that generates arbitrarily large histological images while preserving long-range correlation structural information. Our approach first generates synthetic segmentation masks, subsequently used as conditions for the high-fidelity generative diffusion process. The proposed sampling method can be scaled up to any desired image size while only requiring small patches for fast training. Moreover, it can be parallelized more efficiently than previous large-content generation methods while avoiding tiling artefacts. The training leverages classifier-free guidance to augment a small, sparsely annotated dataset with unlabelled data. Our method alleviates unique challenges in histopathological imaging practice: large-scale information, costly manual annotation, and protective data handling. The biological plausibility of DiffInfinite data is validated in a survey by ten experienced pathologists as well as a downstream segmentation task. Furthermore, the model scores strongly on anti-copying metrics which is beneficial for the protection of patient data.
Leveraging weak complementary labels to improve semantic segmentation of hepatocellular carcinoma and cholangiocarcinoma in H&E-stained slides
Feb 03, 2023



In this paper, we present a deep learning segmentation approach to classify and quantify the two most prevalent primary liver cancers - hepatocellular carcinoma and intrahepatic cholangiocarcinoma - from hematoxylin and eosin (H&E) stained whole slide images. While semantic segmentation of medical images typically requires costly pixel-level annotations by domain experts, there often exists additional information which is routinely obtained in clinical diagnostics but rarely utilized for model training. We propose to leverage such weak information from patient diagnoses by deriving complementary labels that indicate to which class a sample cannot belong to. To integrate these labels, we formulate a complementary loss for segmentation. Motivated by the medical application, we demonstrate for general segmentation tasks that including additional patches with solely weak complementary labels during model training can significantly improve the predictive performance and robustness of a model. On the task of diagnostic differentiation between hepatocellular carcinoma and intrahepatic cholangiocarcinoma, we achieve a balanced accuracy of 0.91 (CI 95%: 0.86 - 0.95) at case level for 165 hold-out patients. Furthermore, we also show that leveraging complementary labels improves the robustness of segmentation and increases performance at case level.